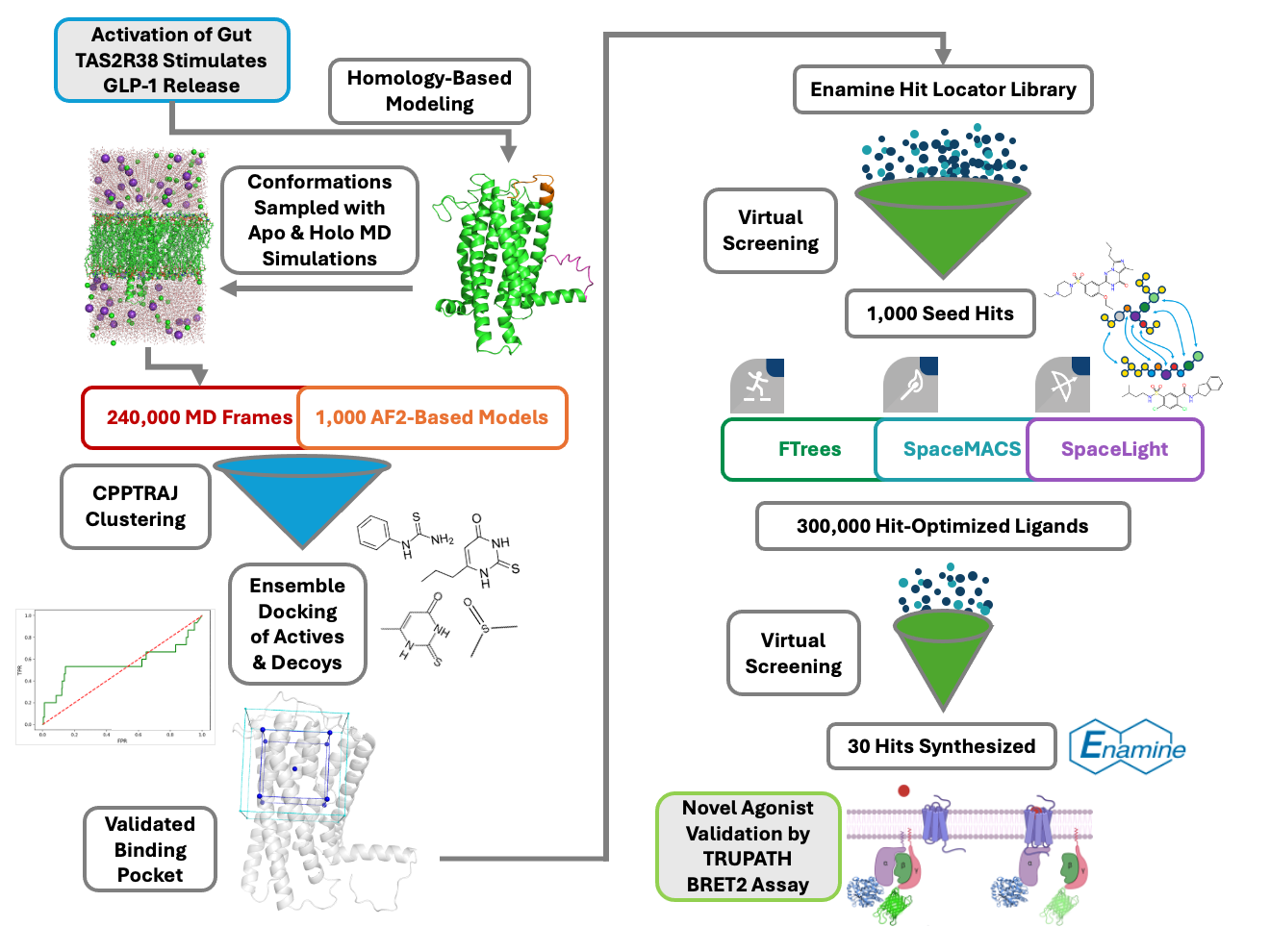
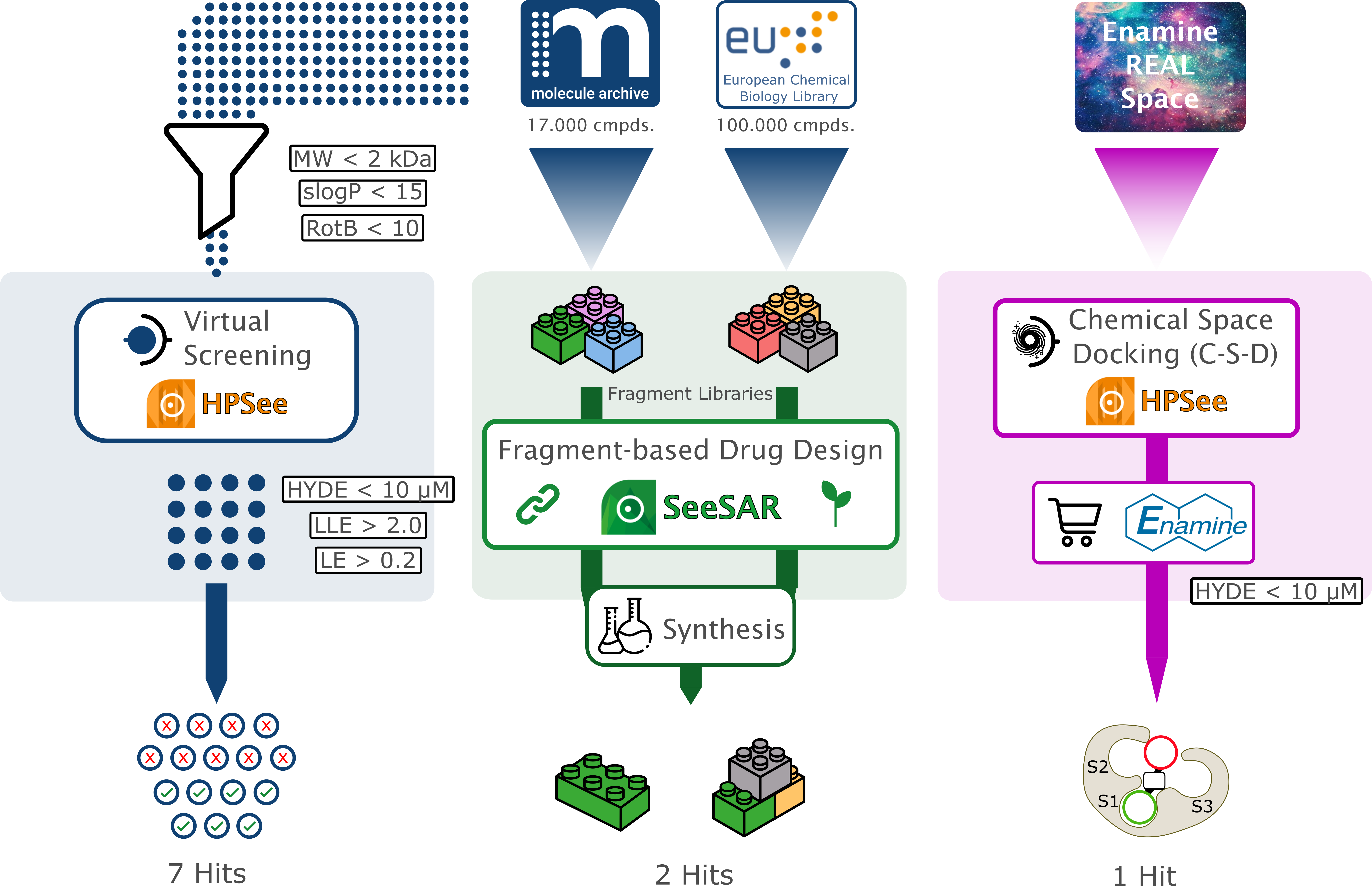
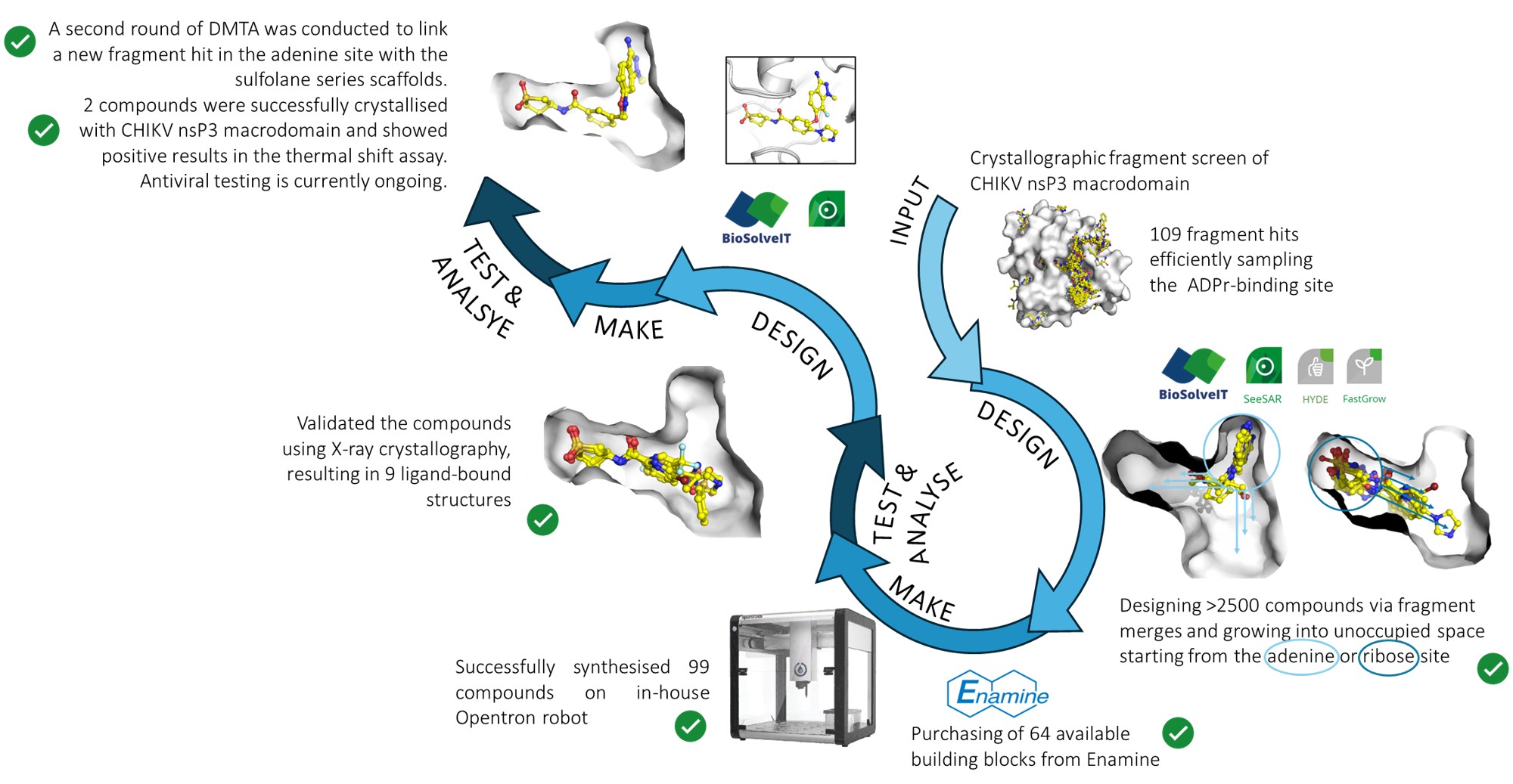
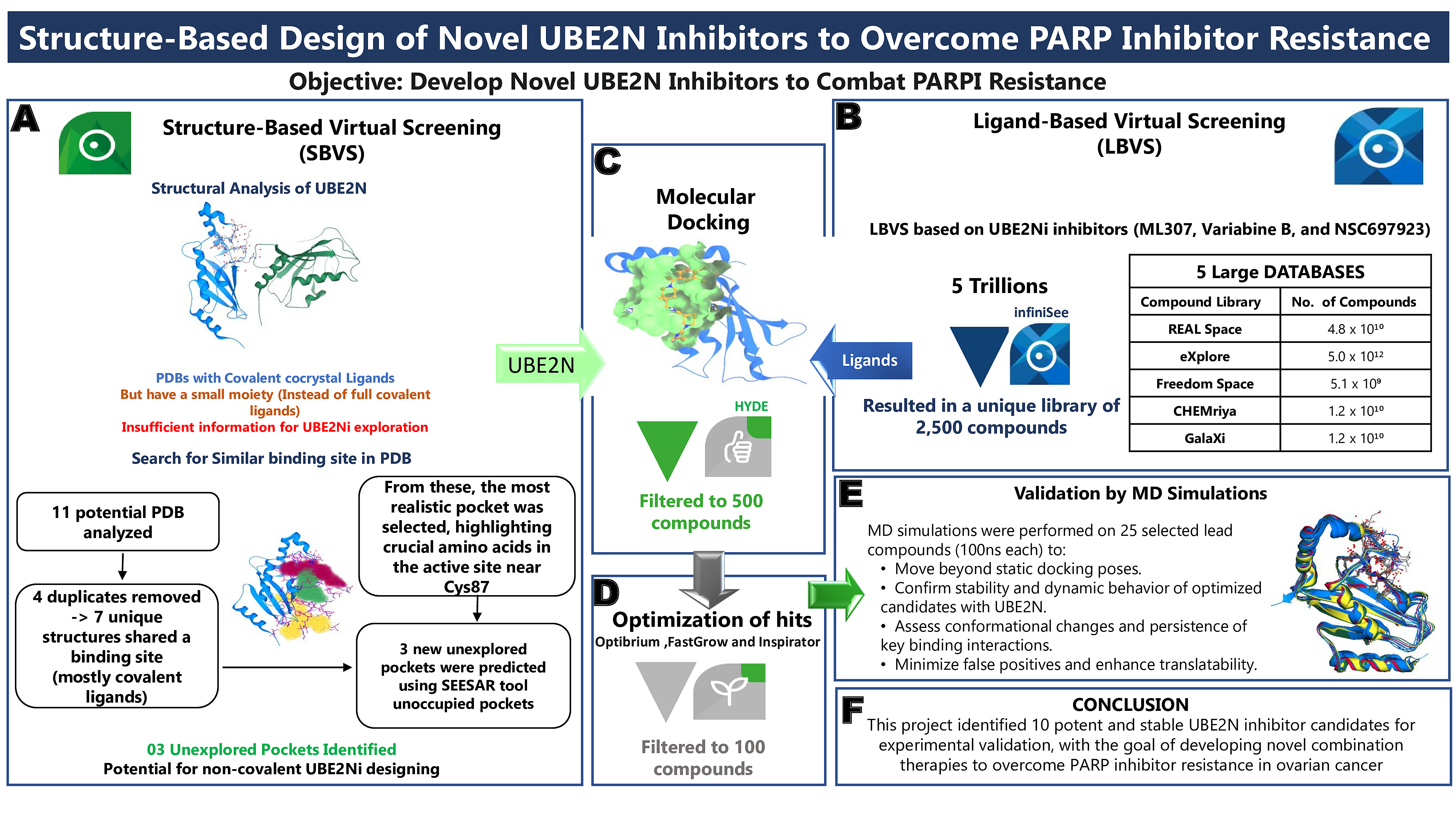
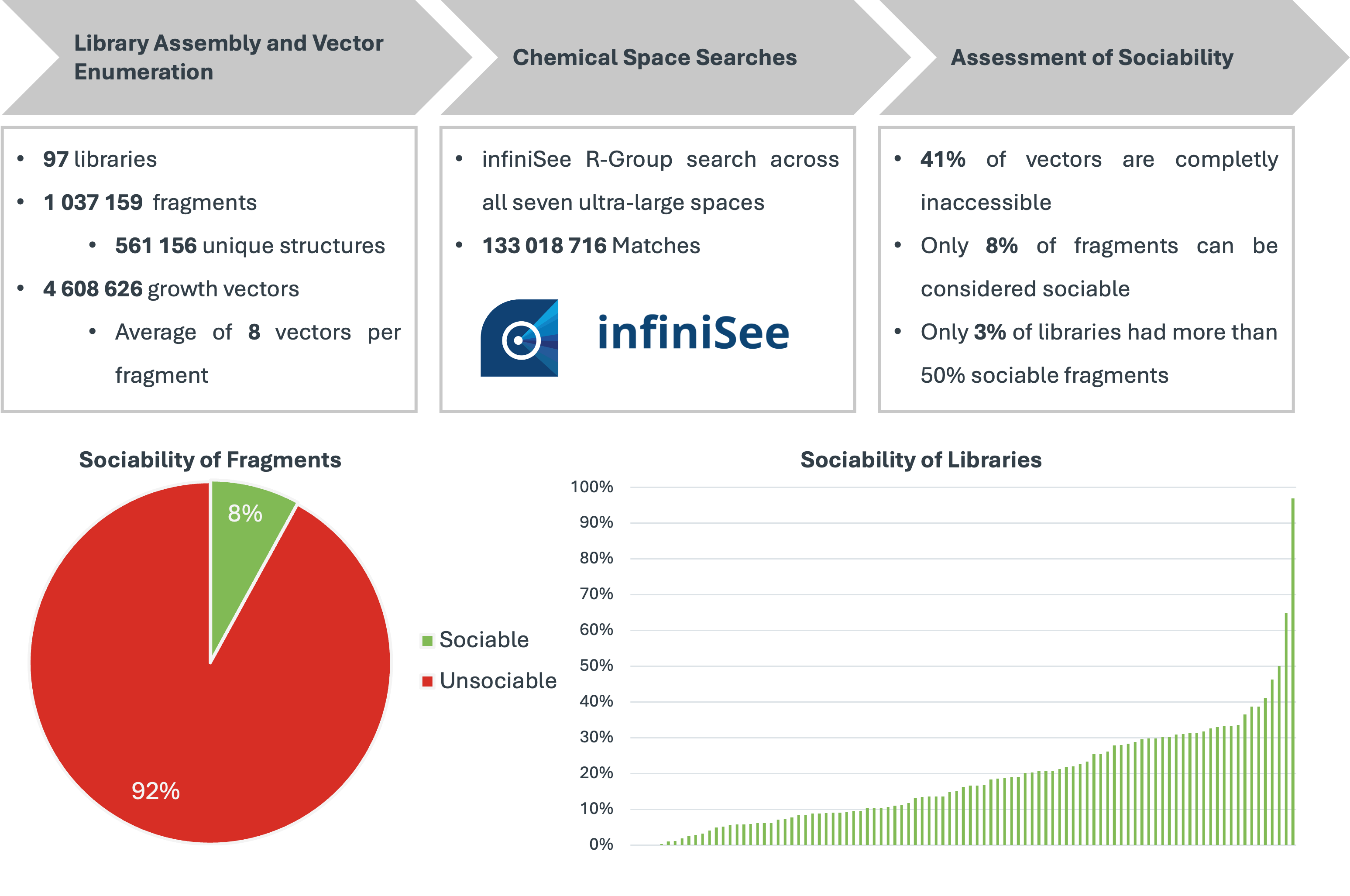
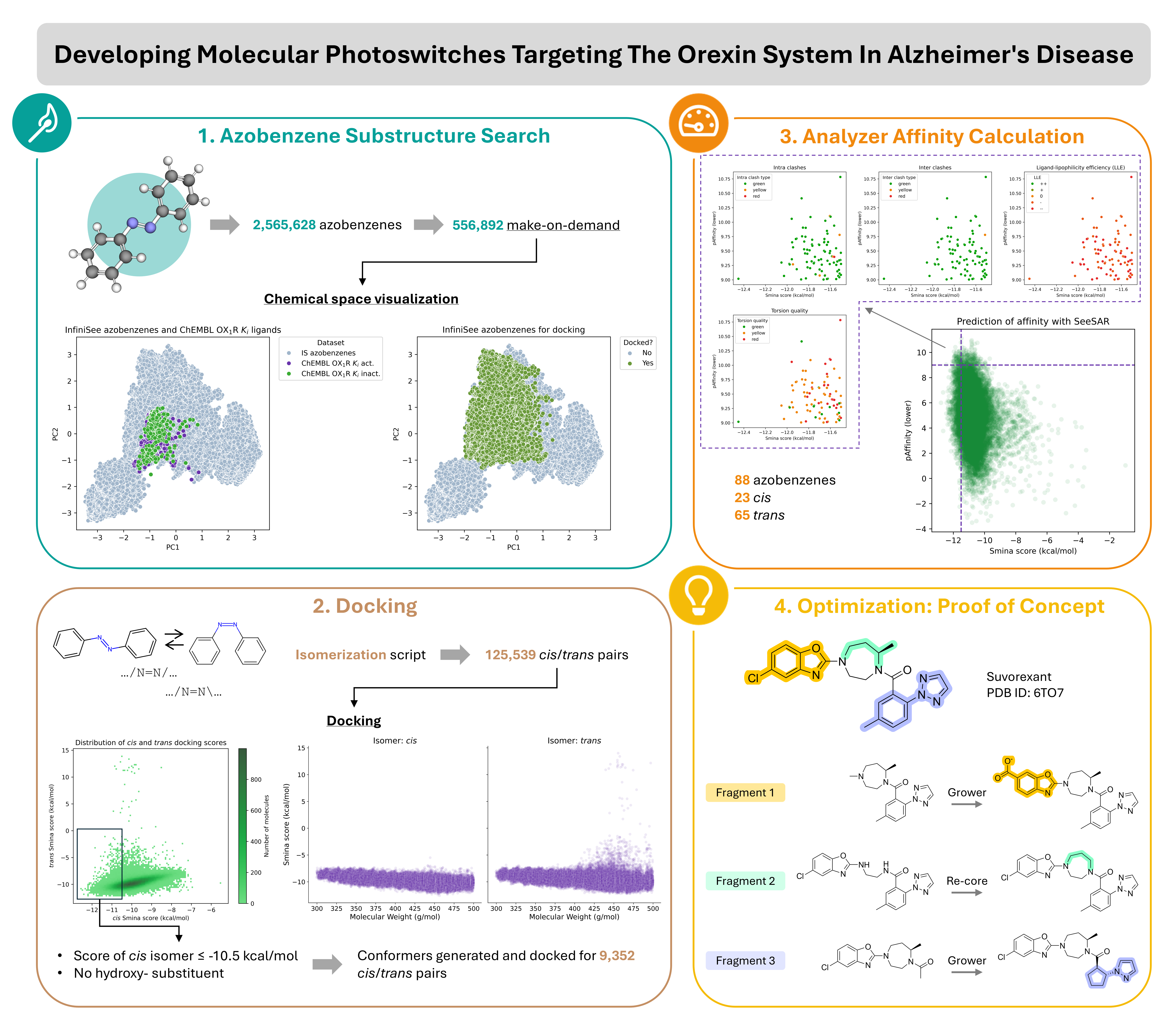
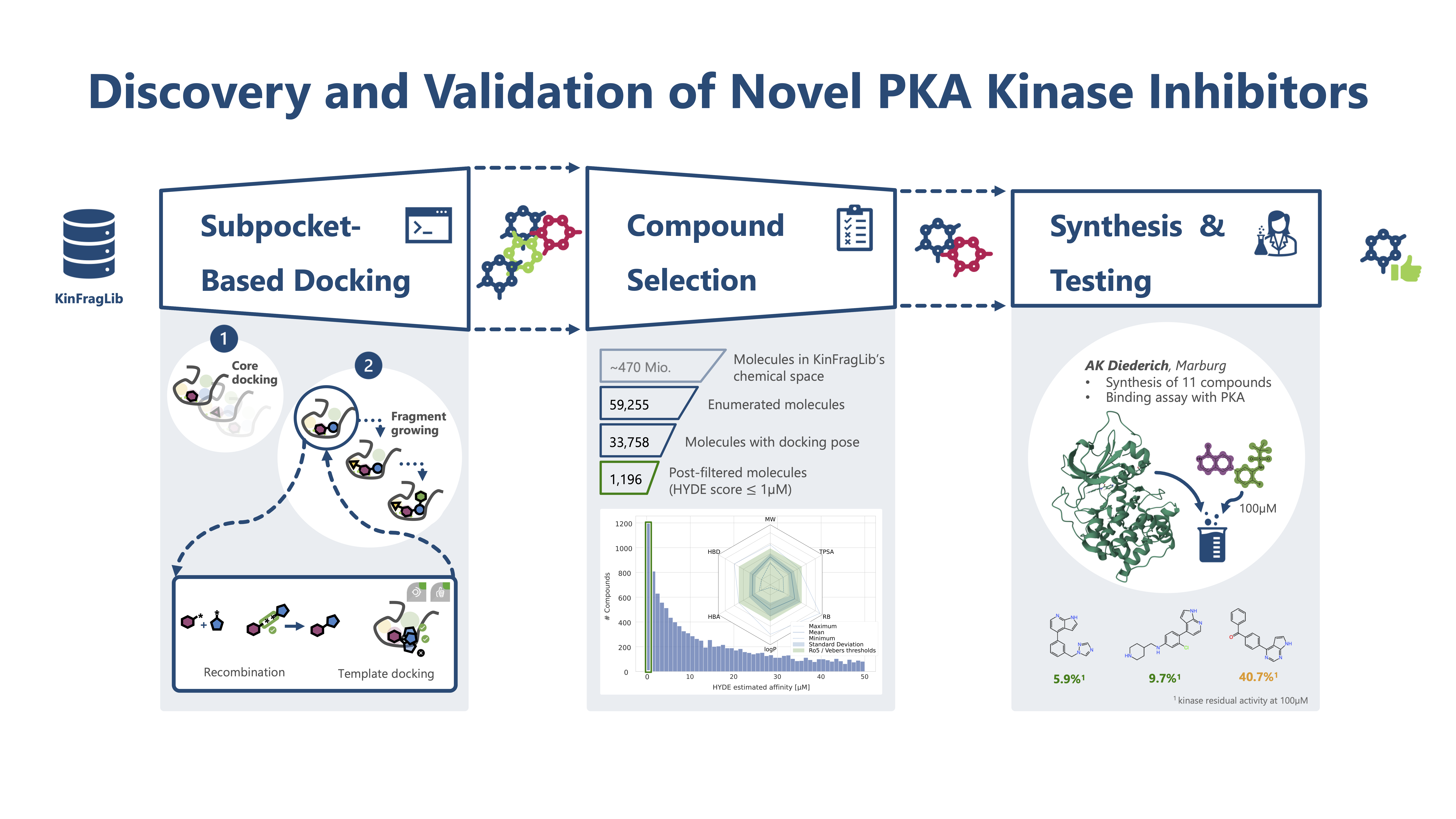
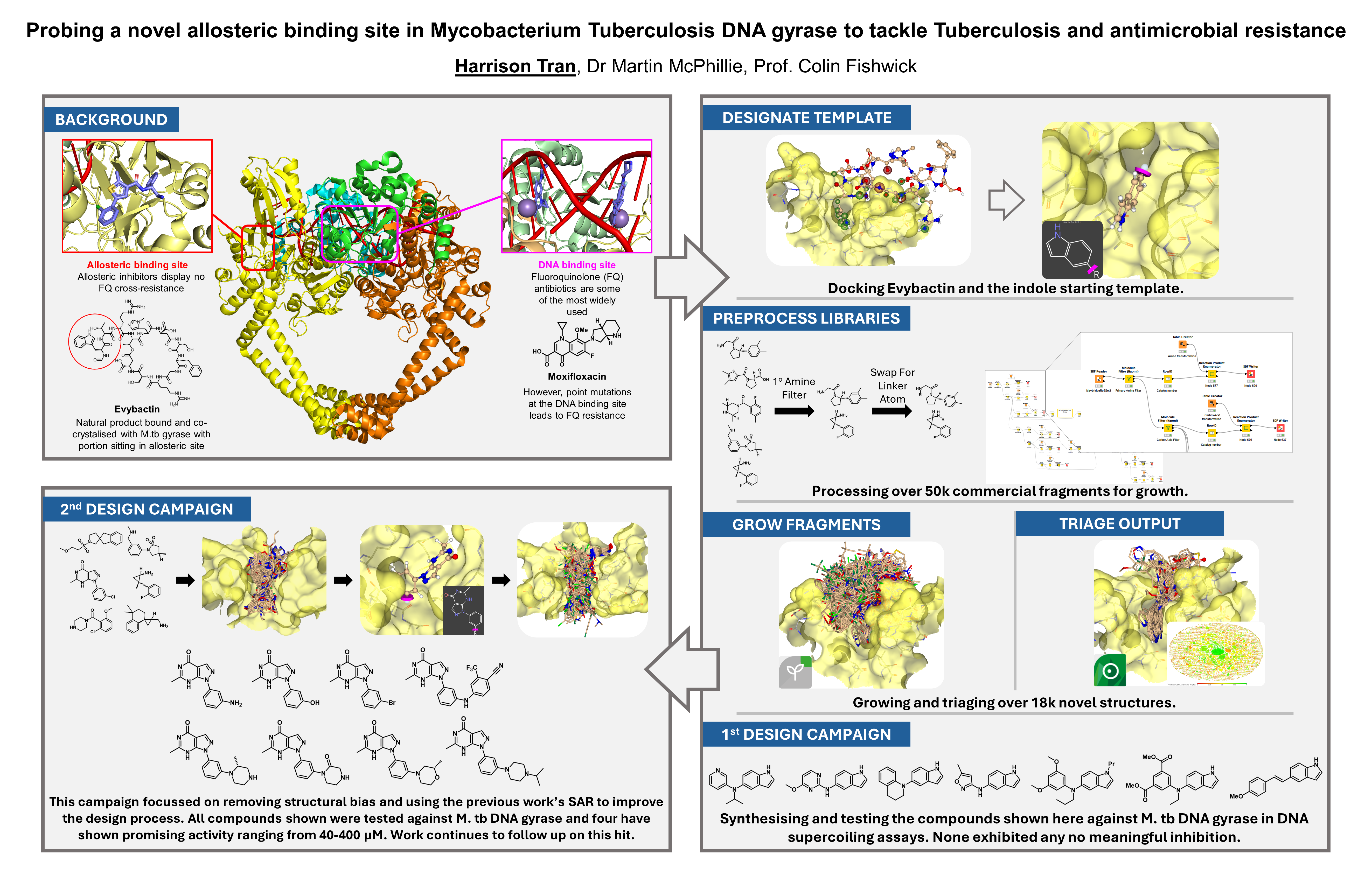
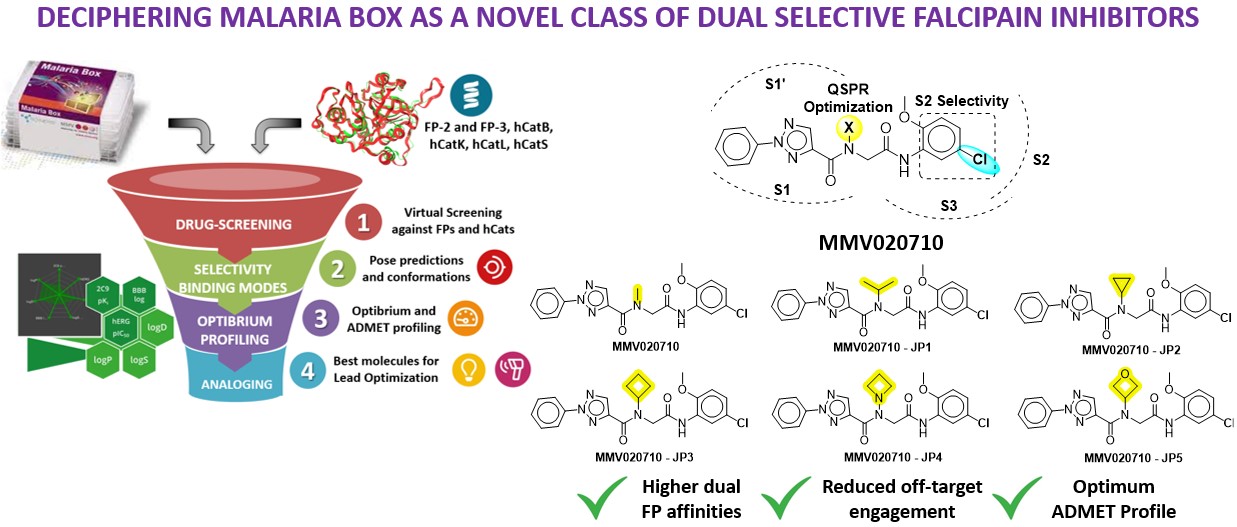
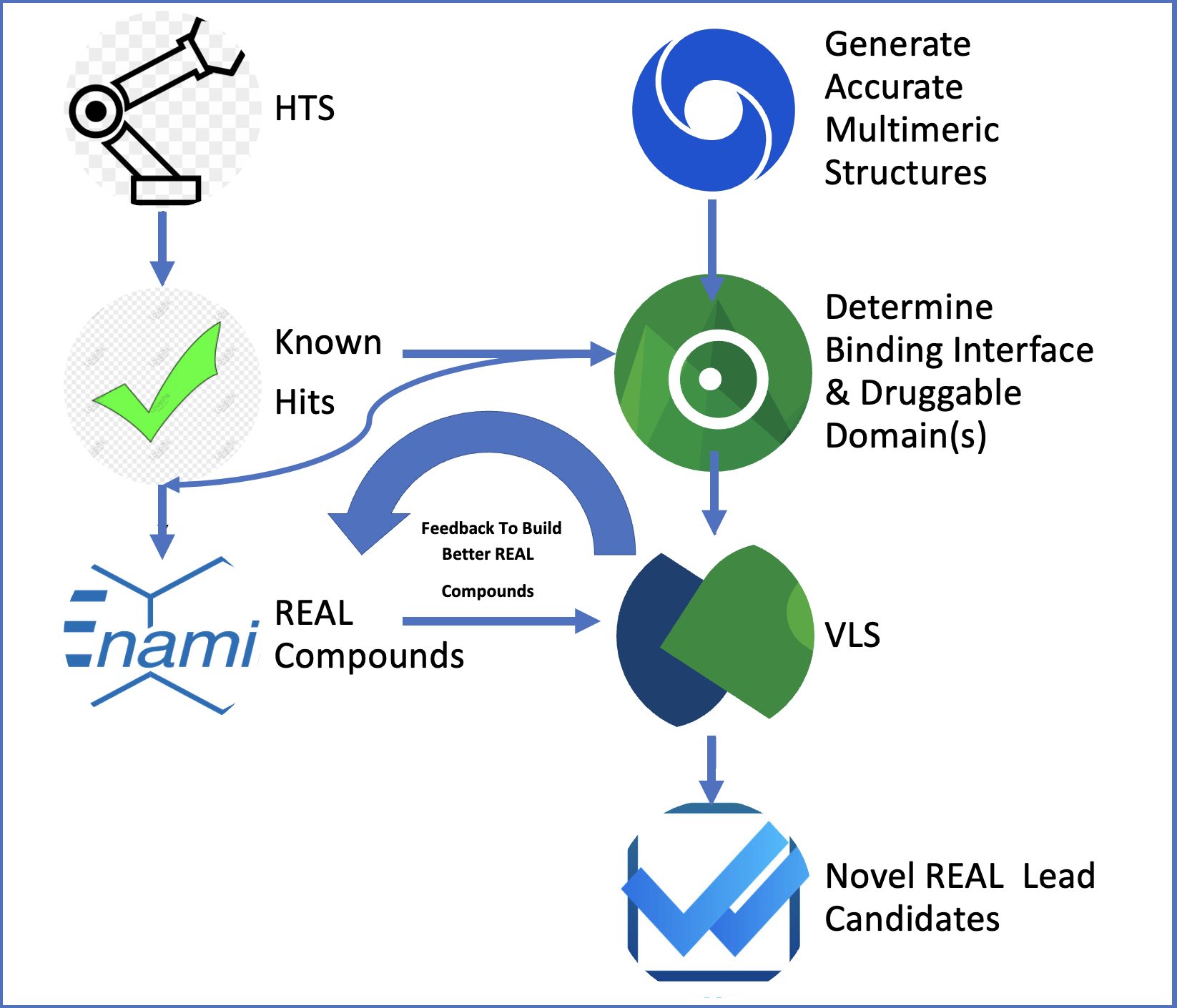
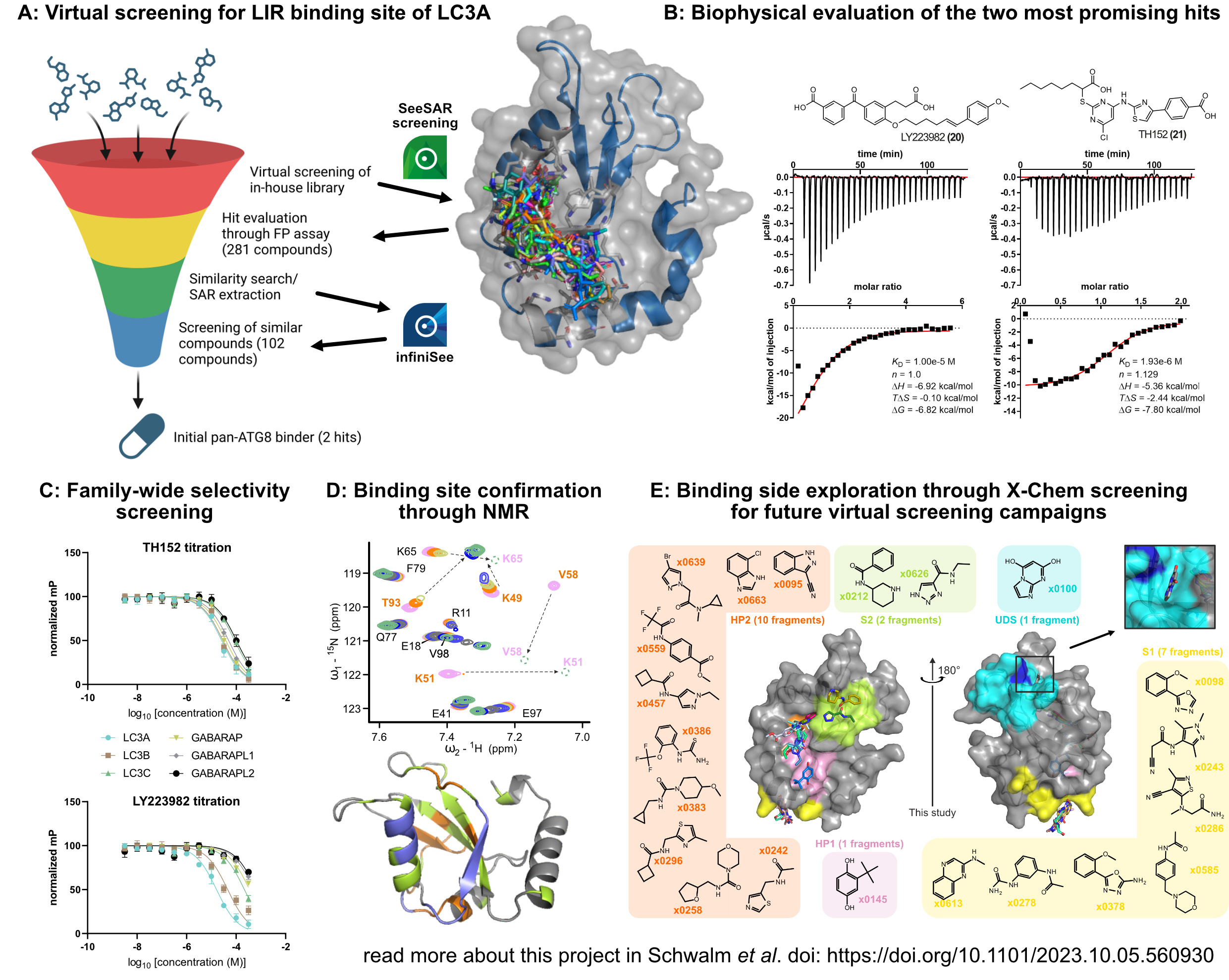
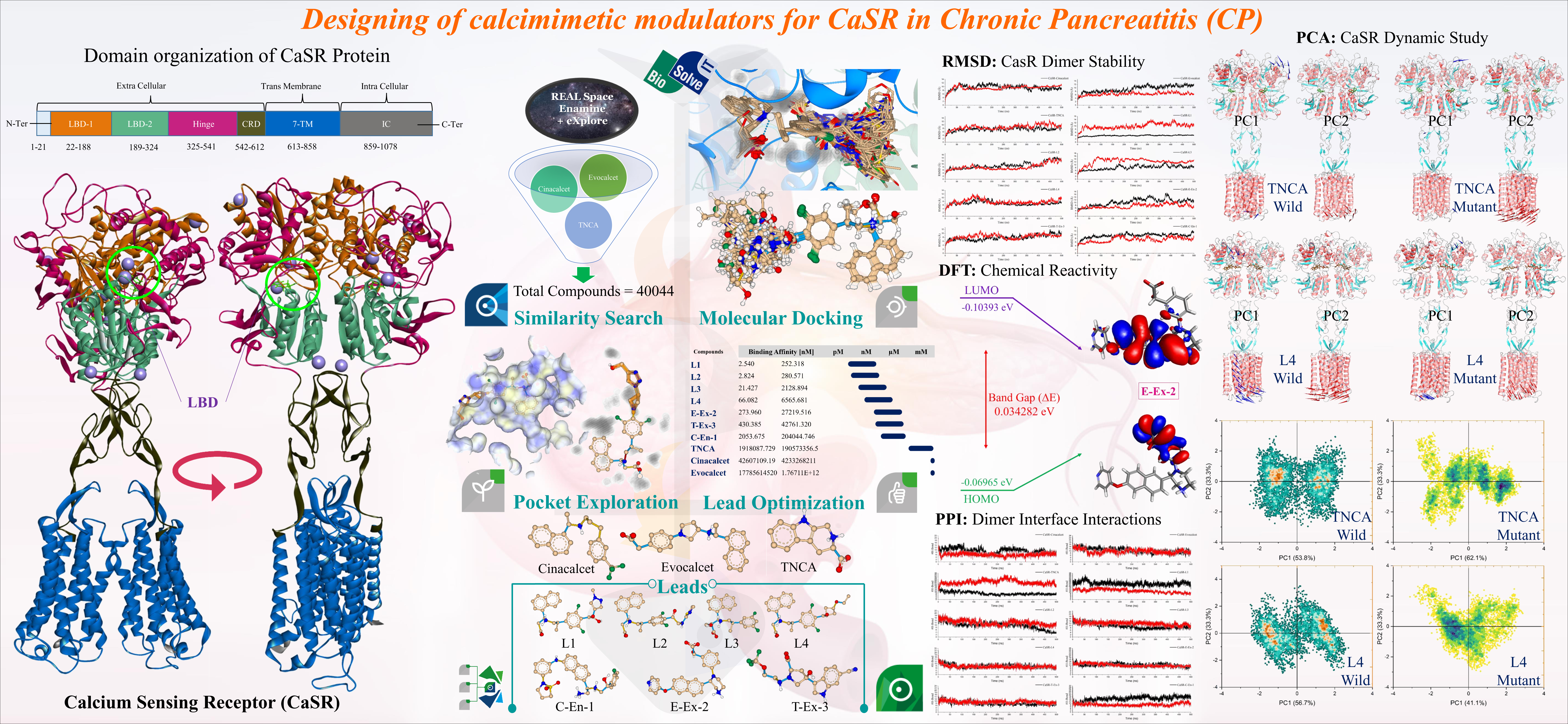
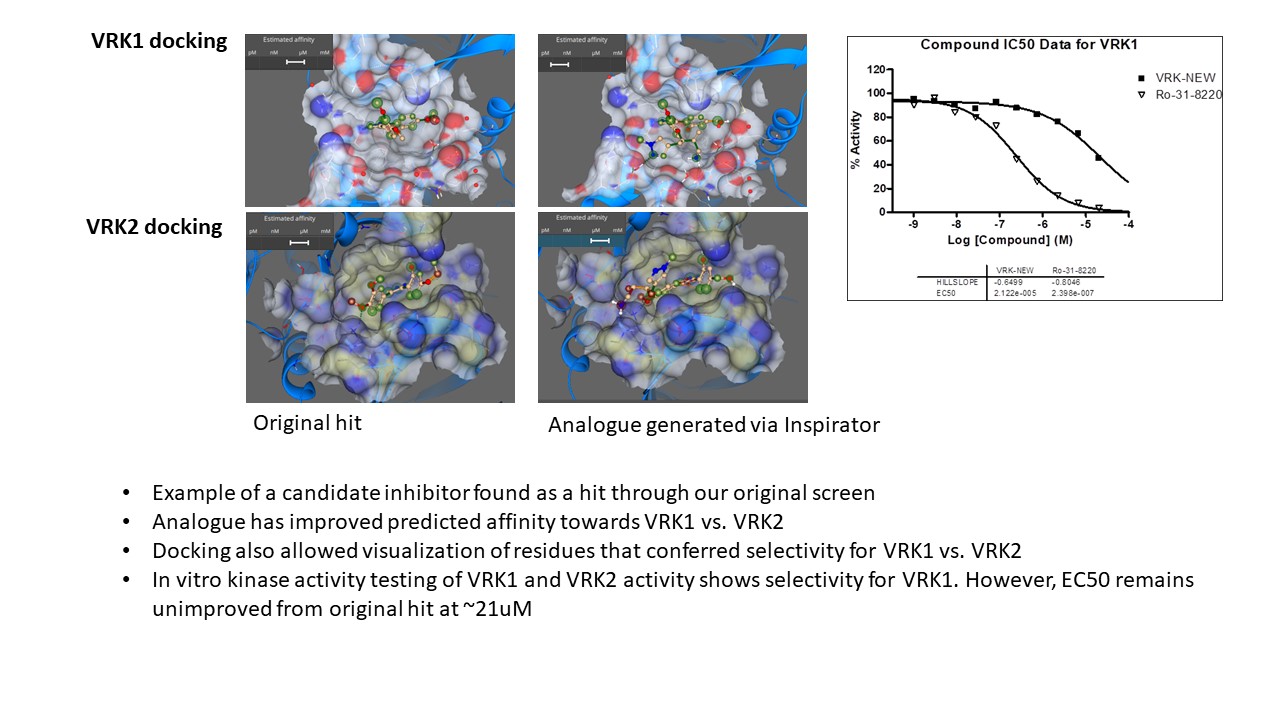
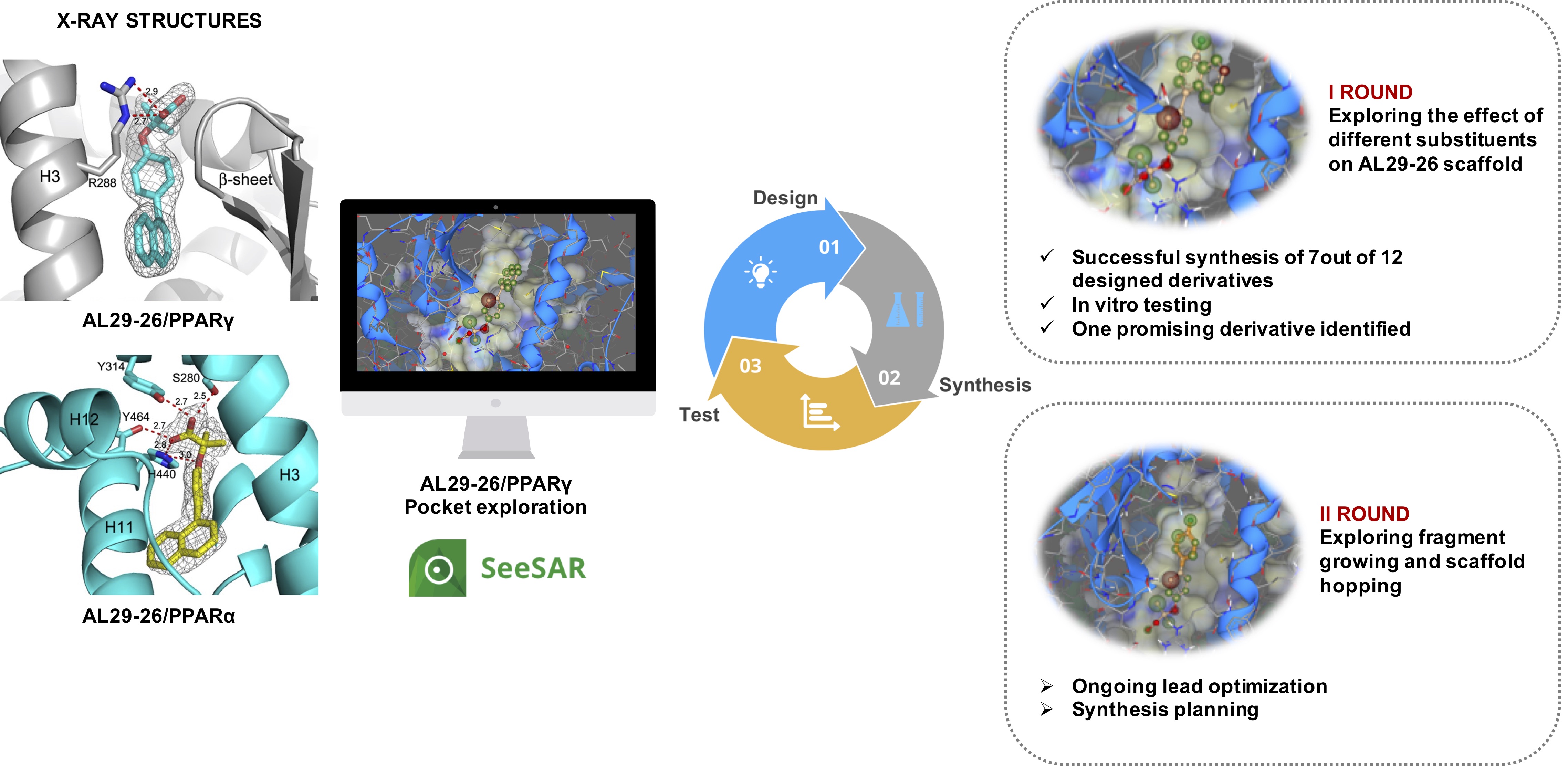
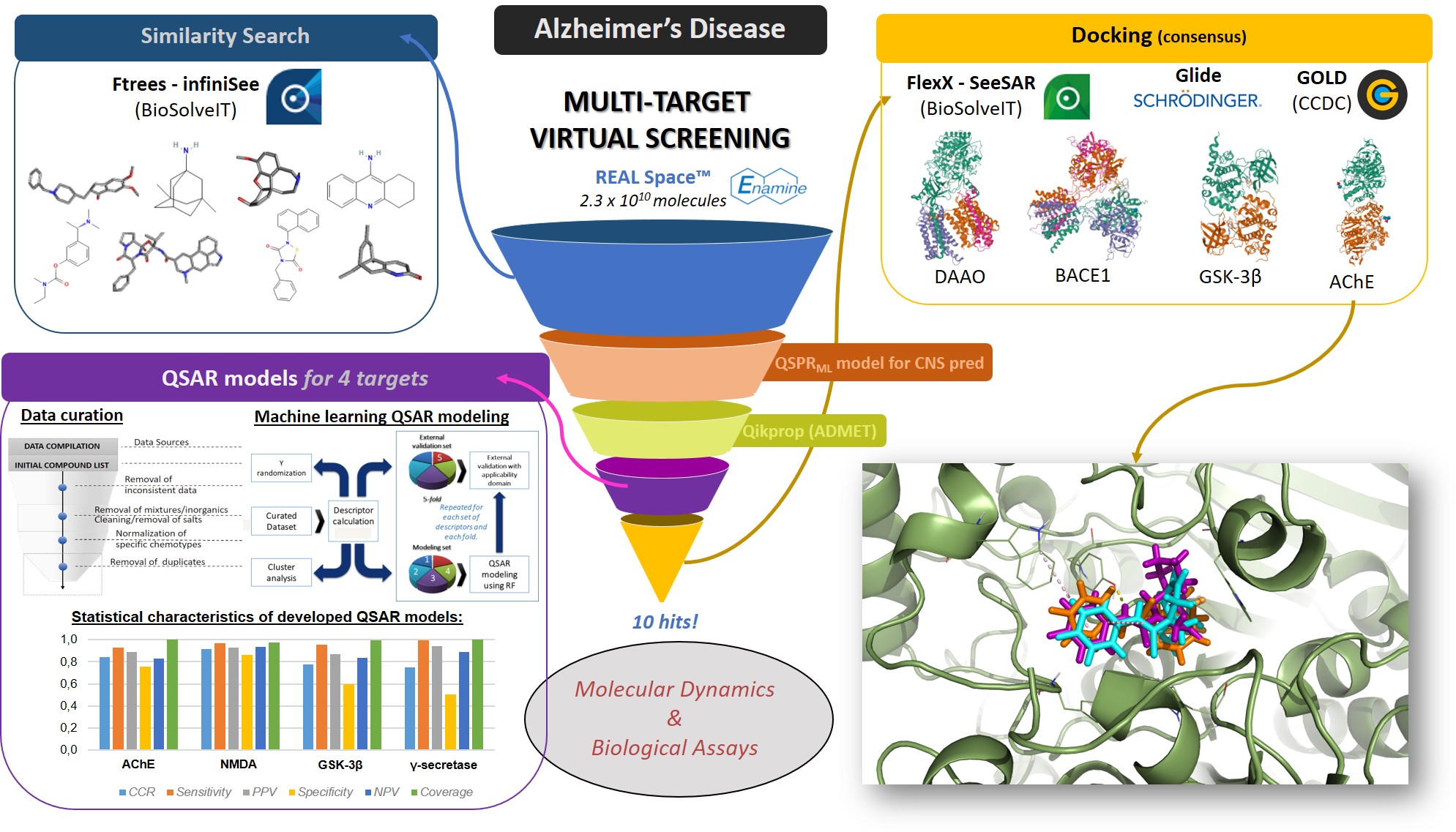
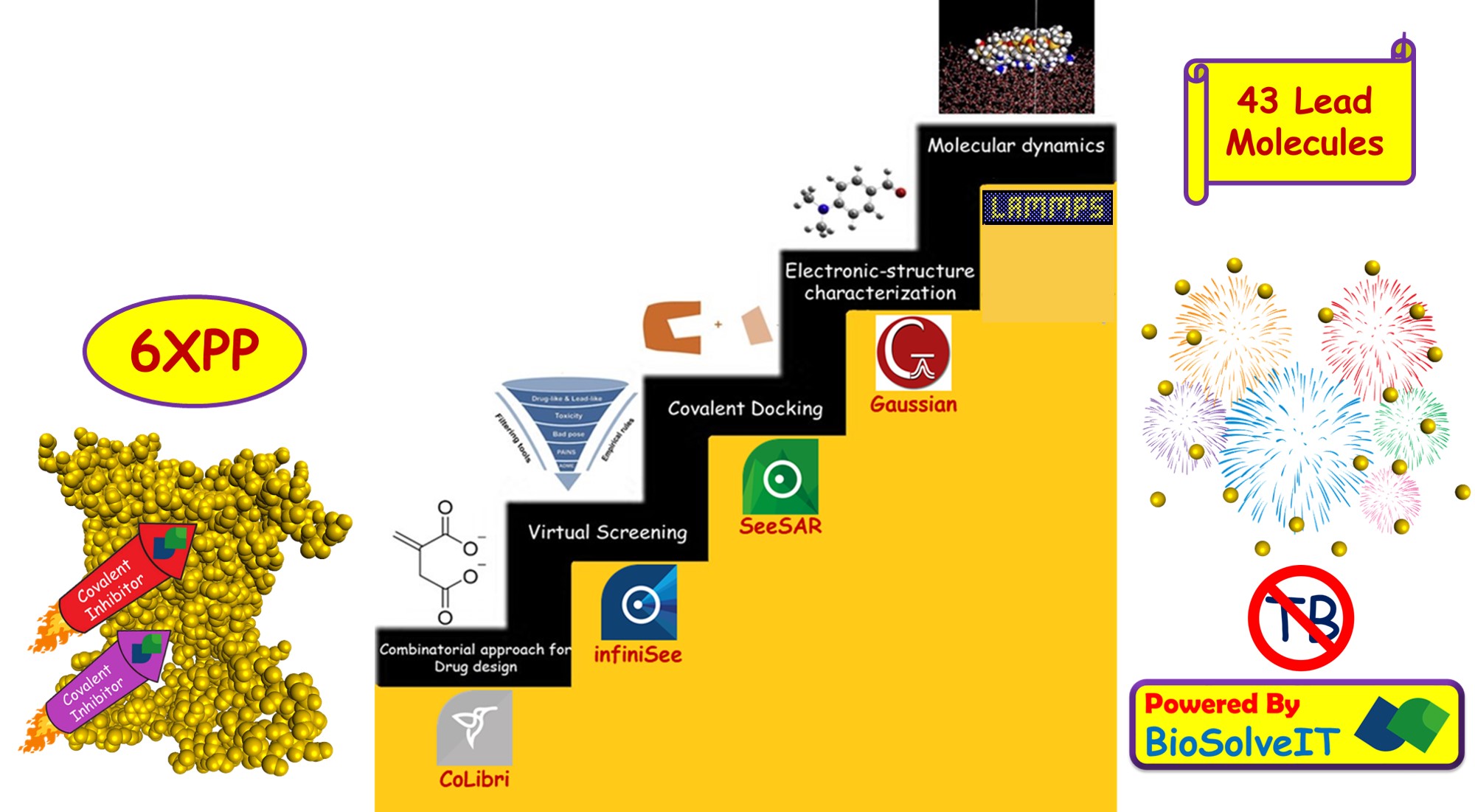
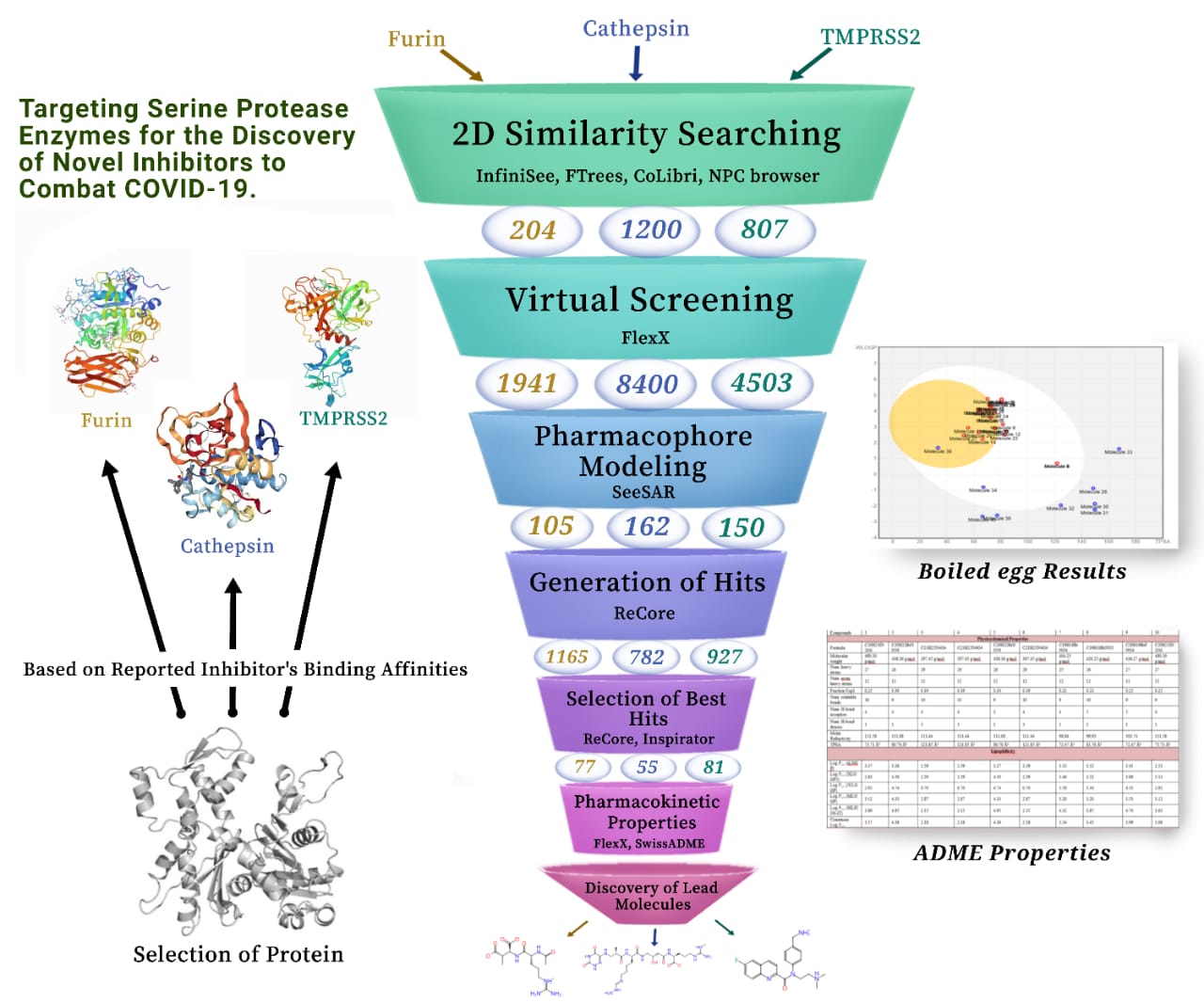
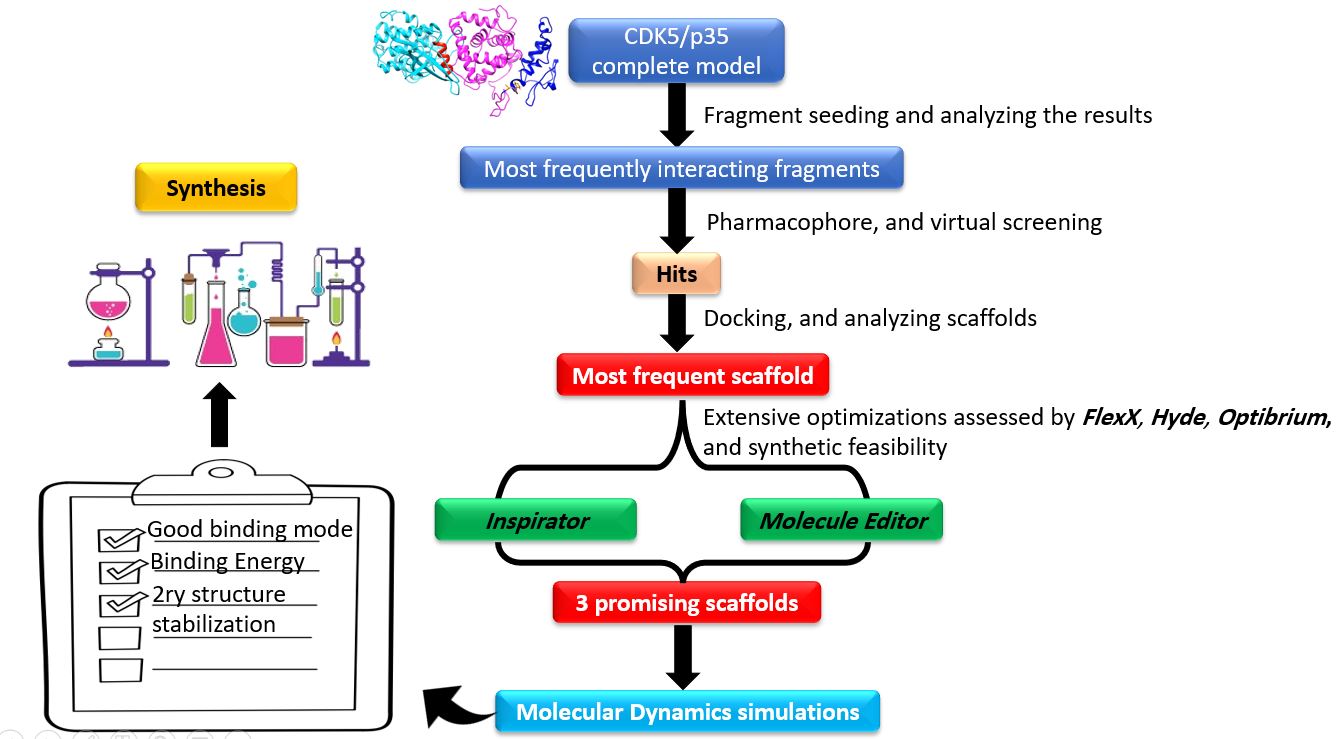
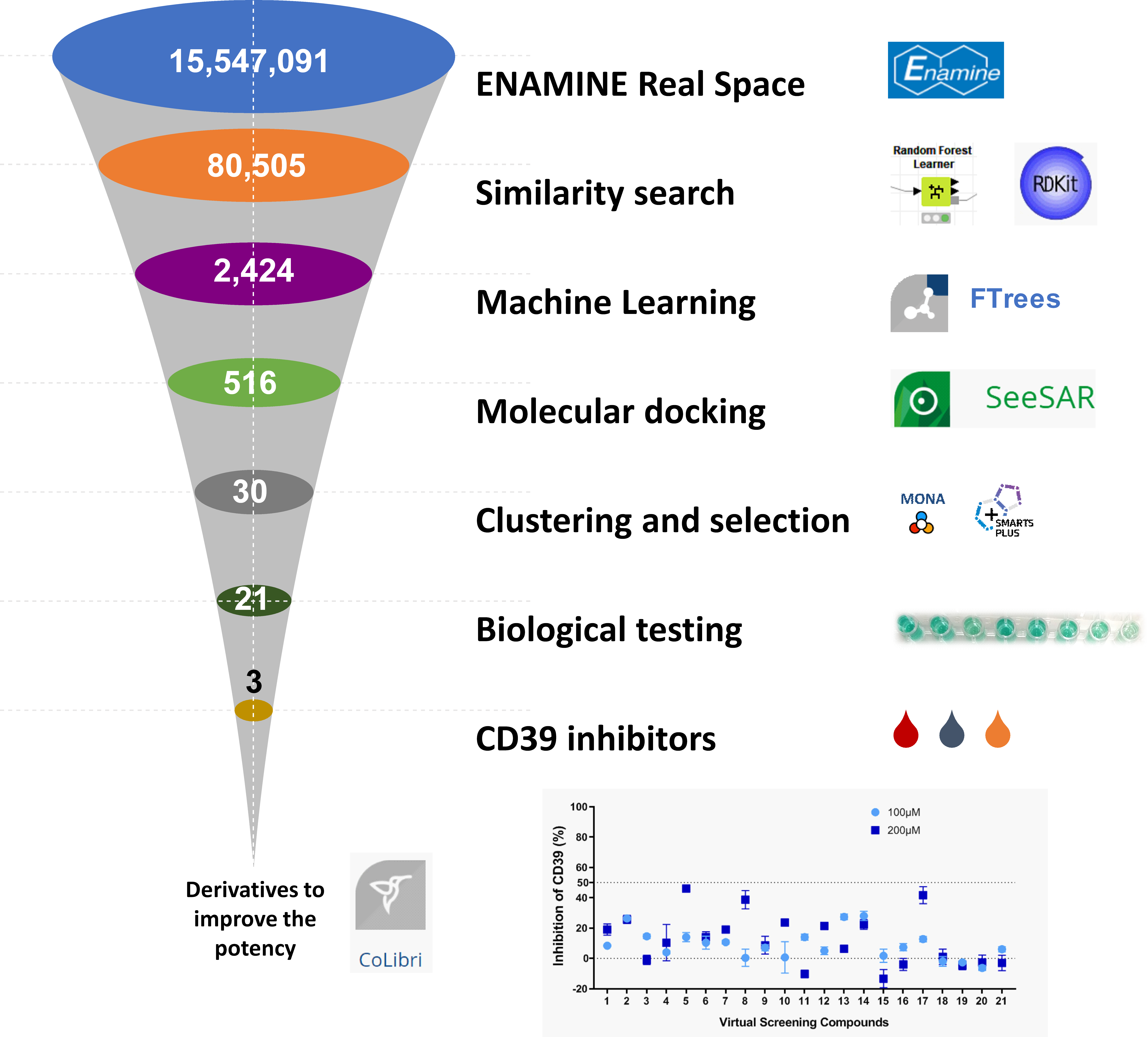
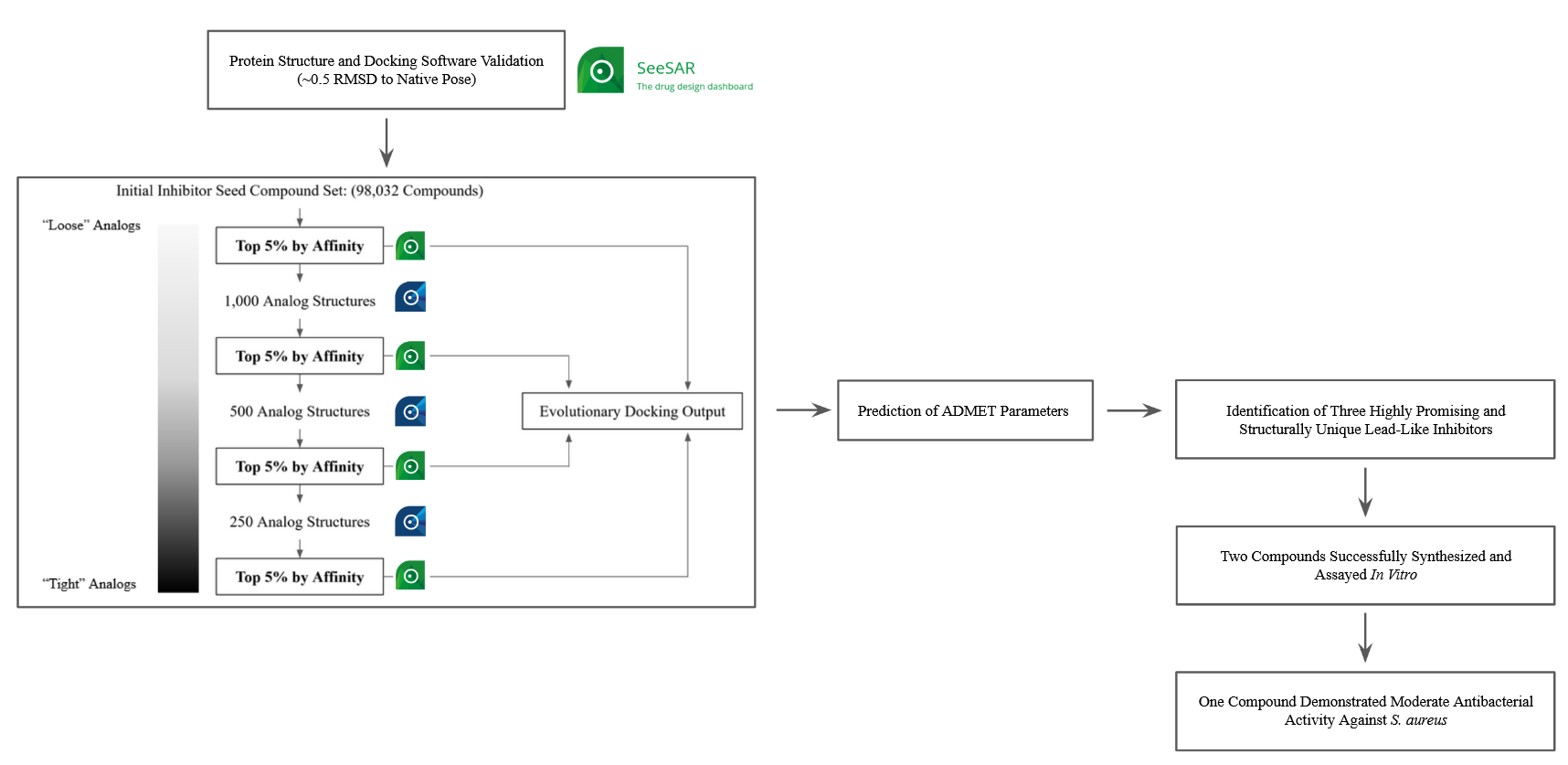
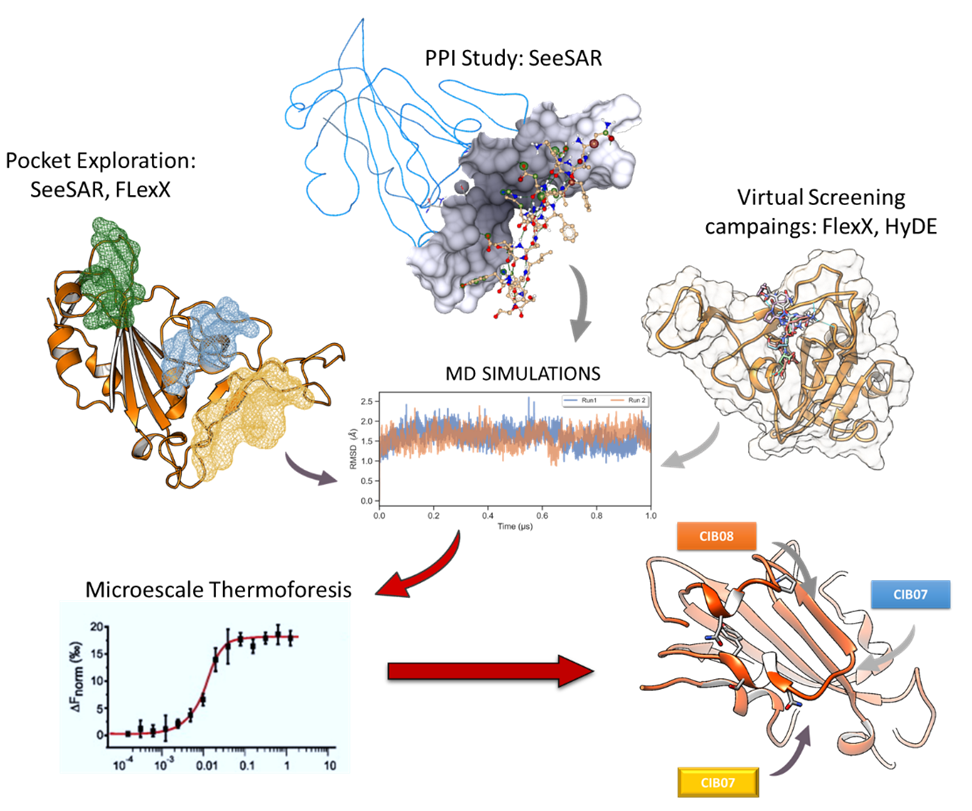
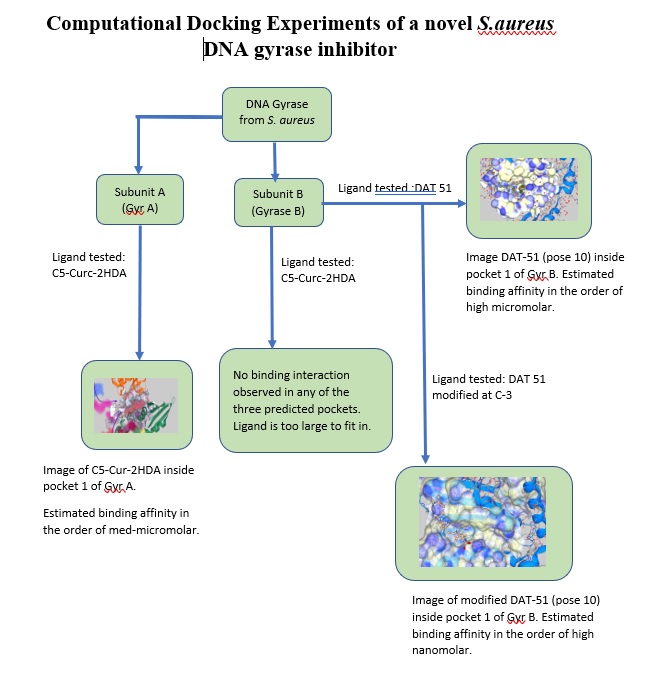
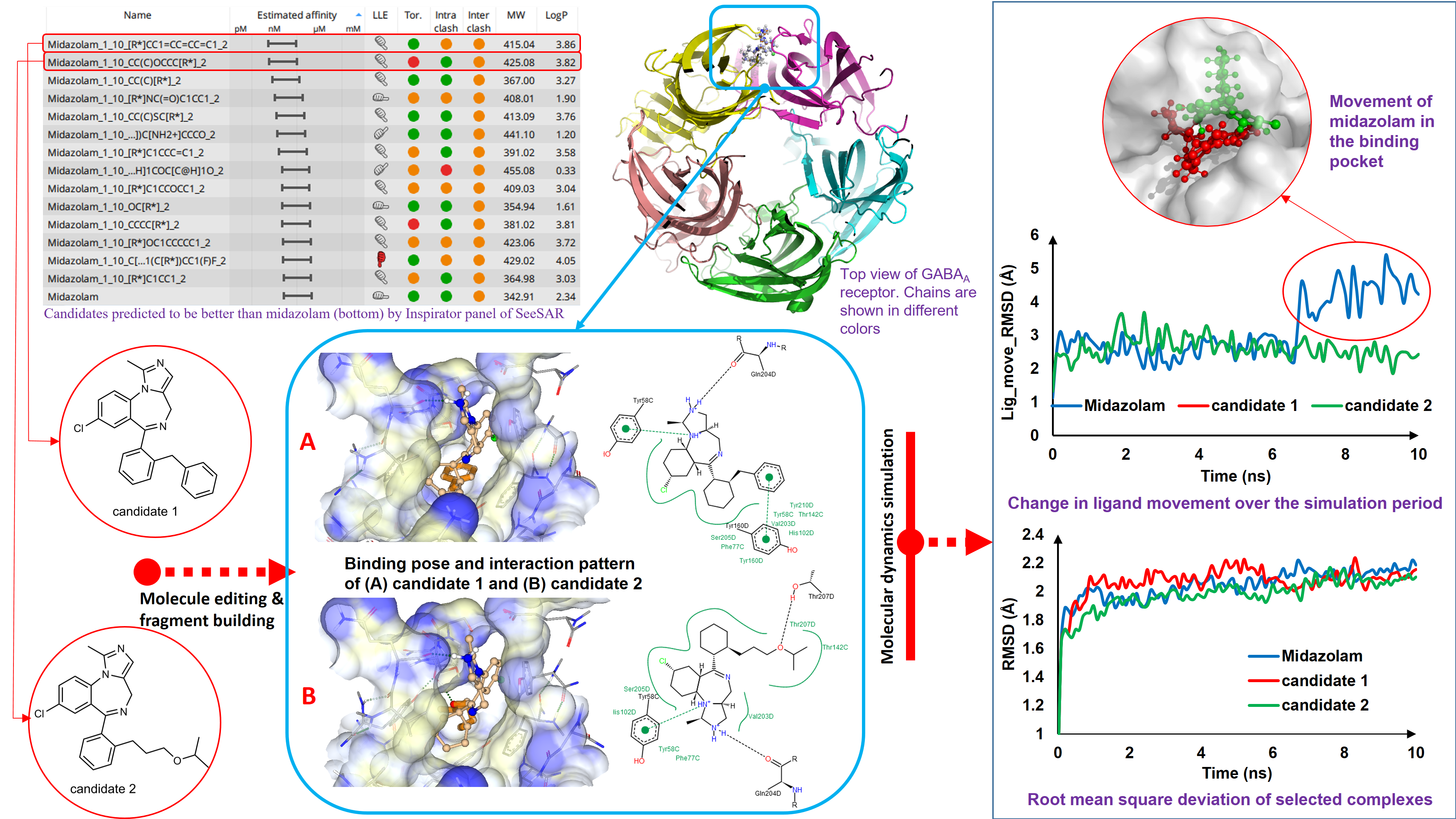
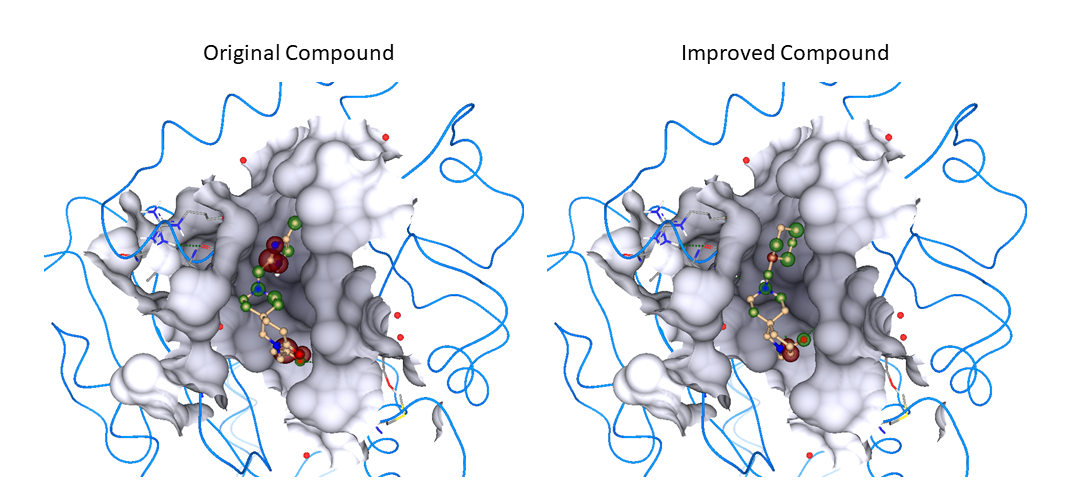
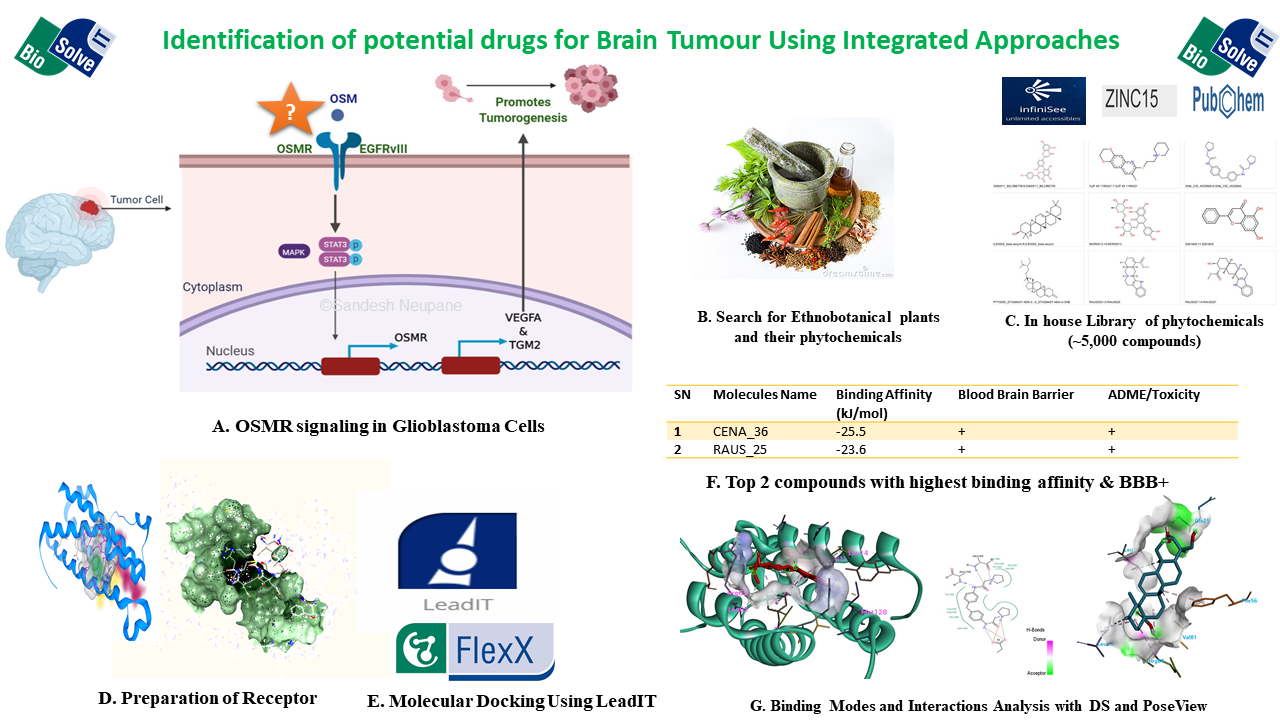
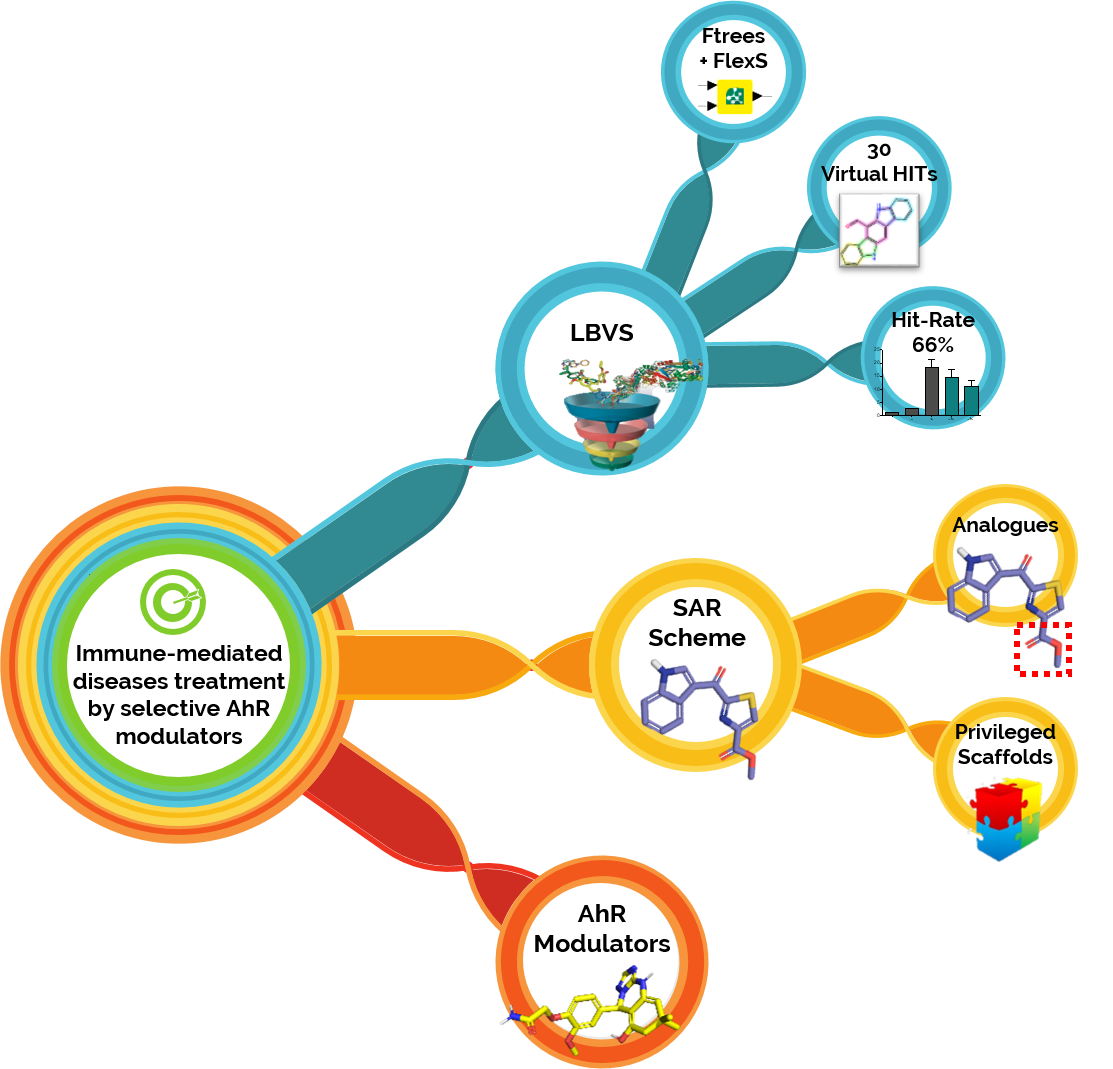
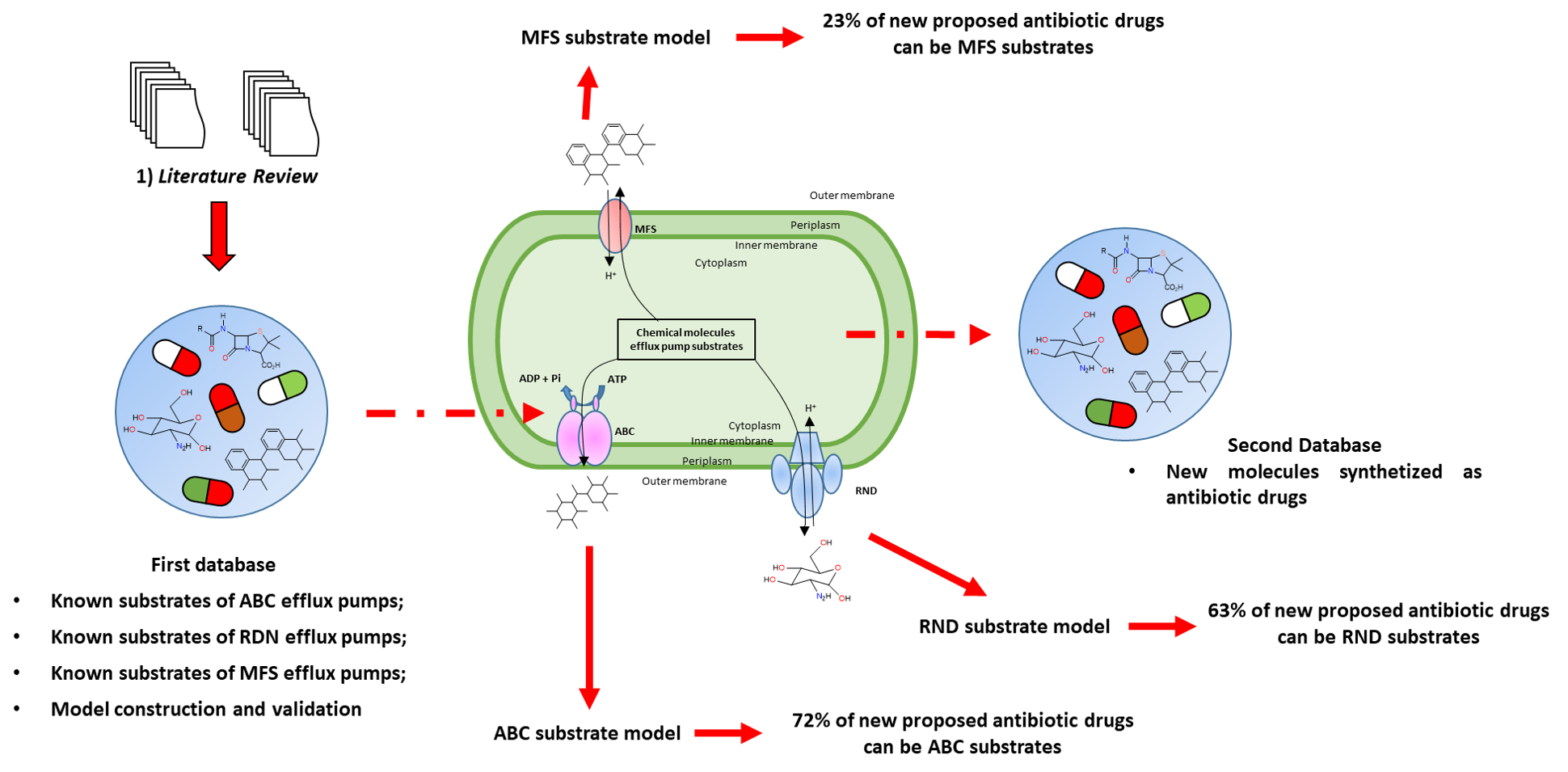
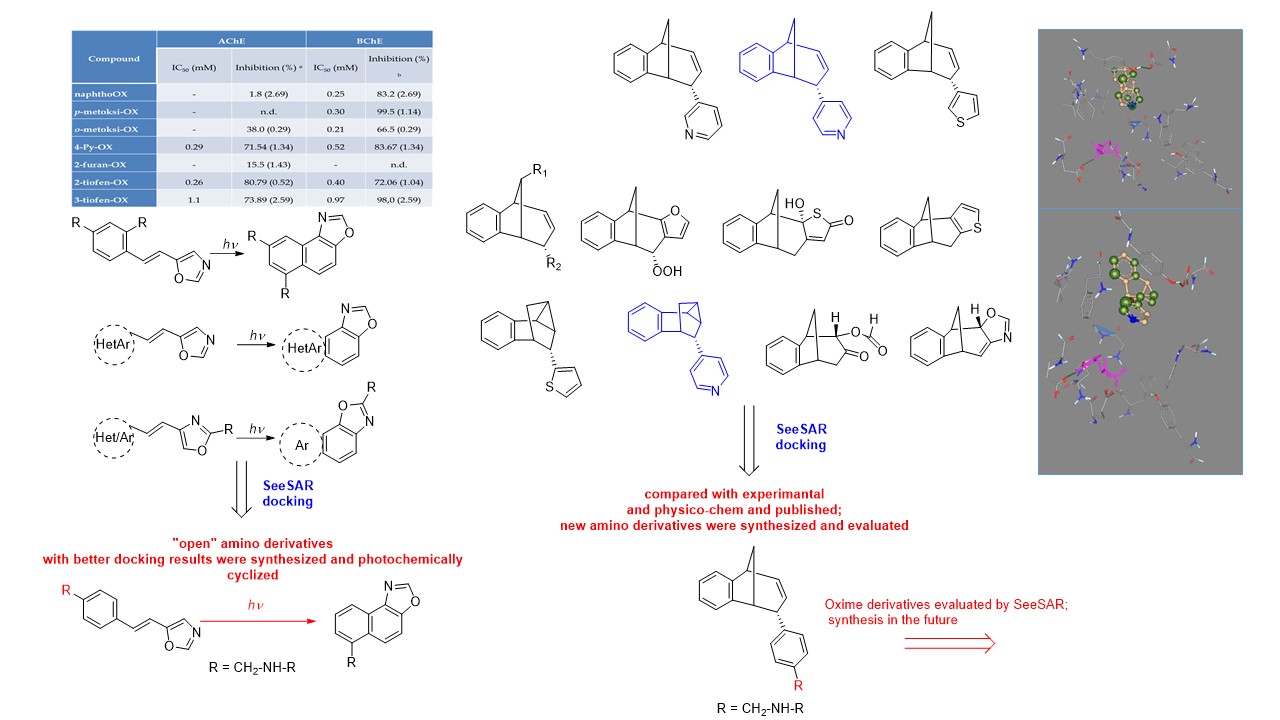
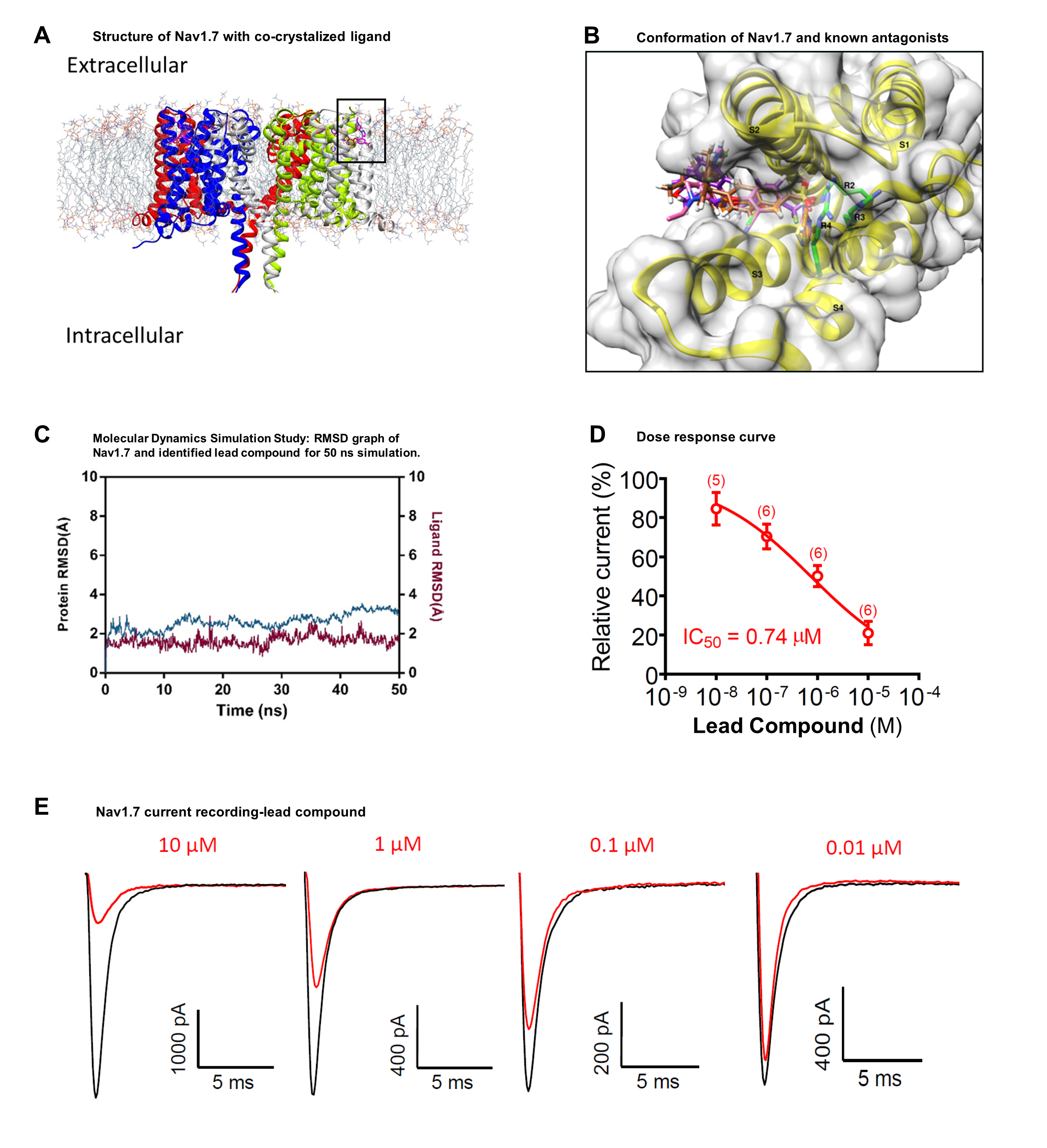
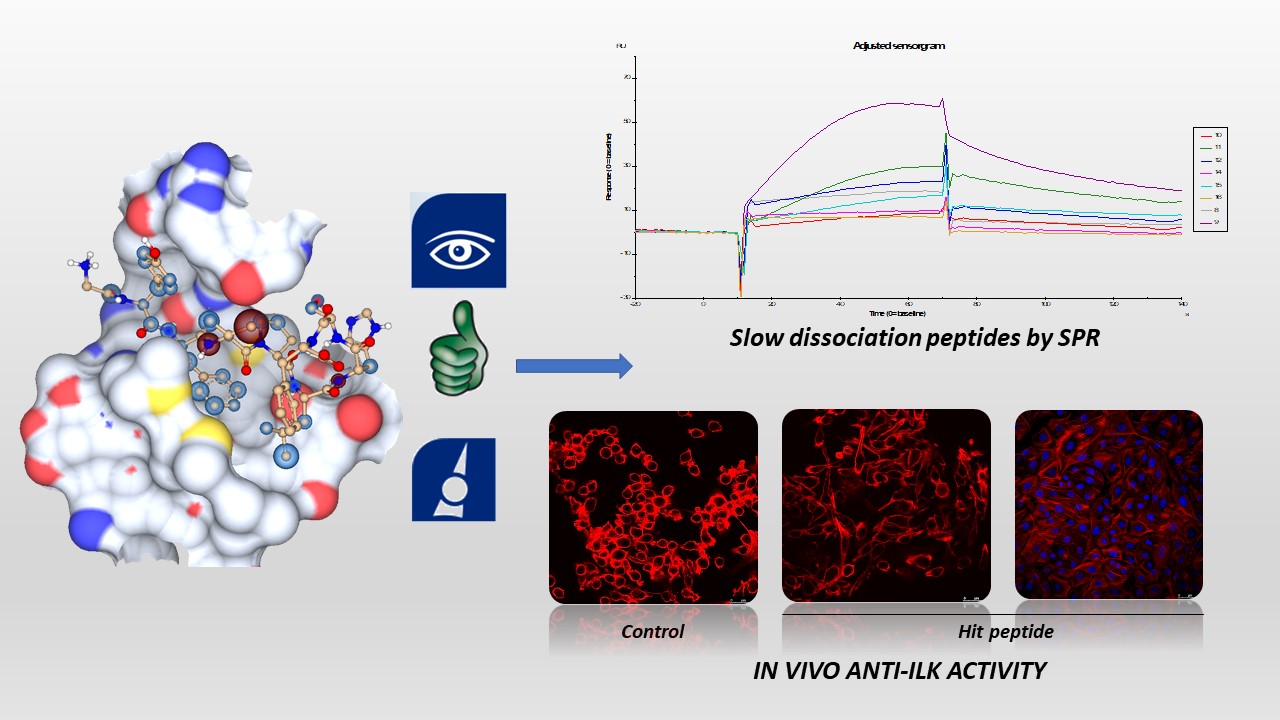
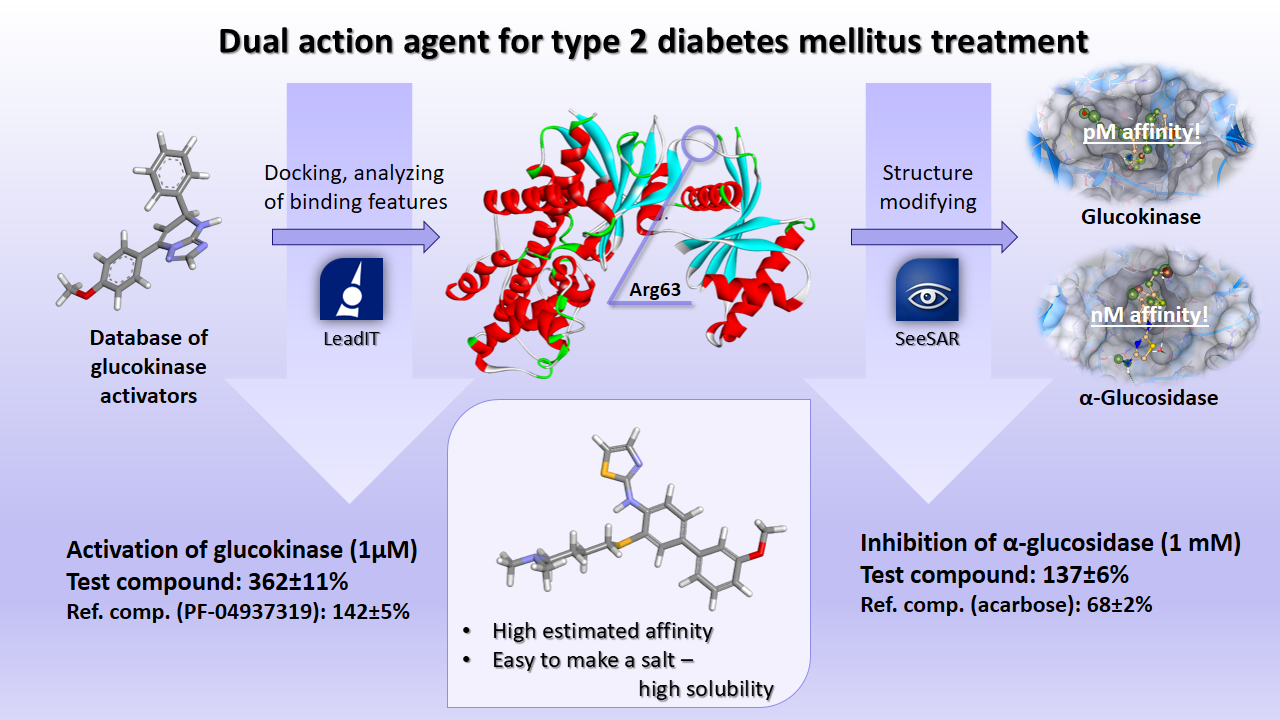
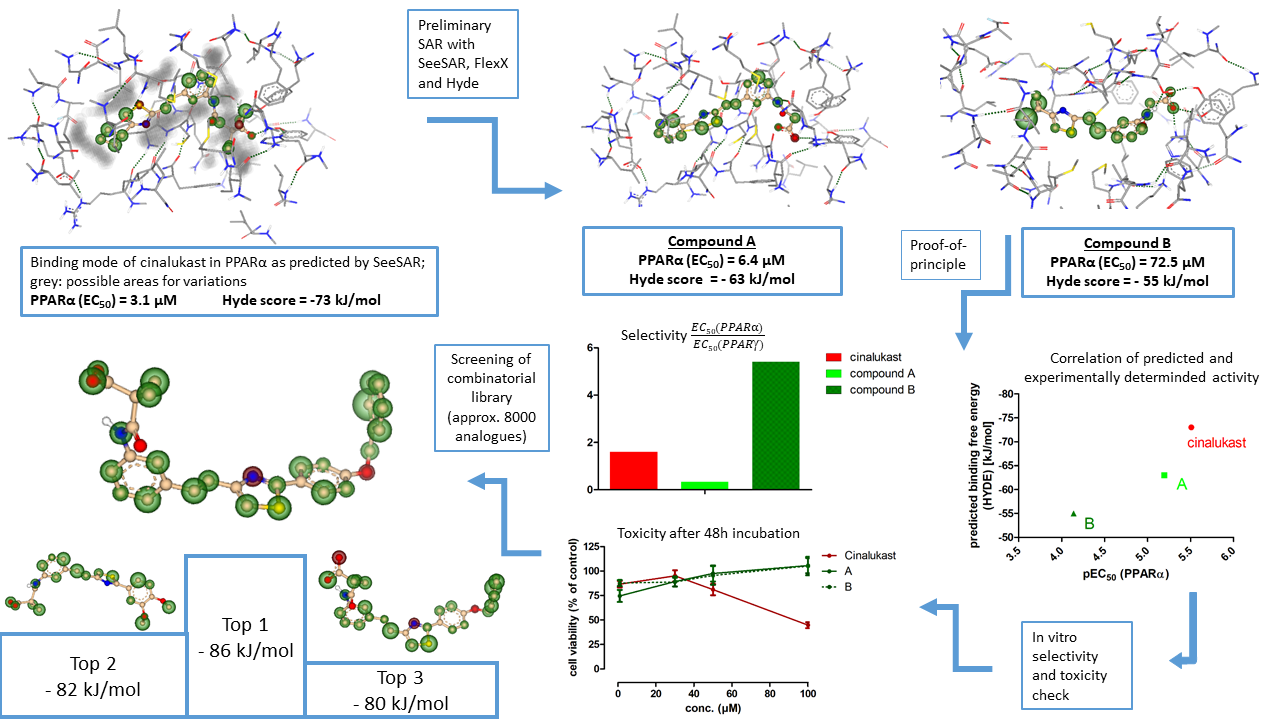
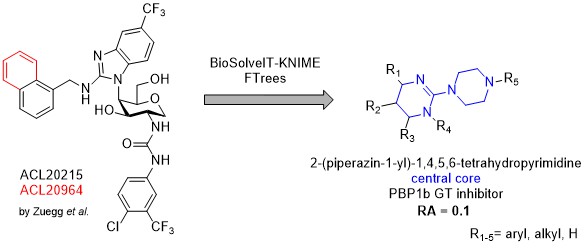
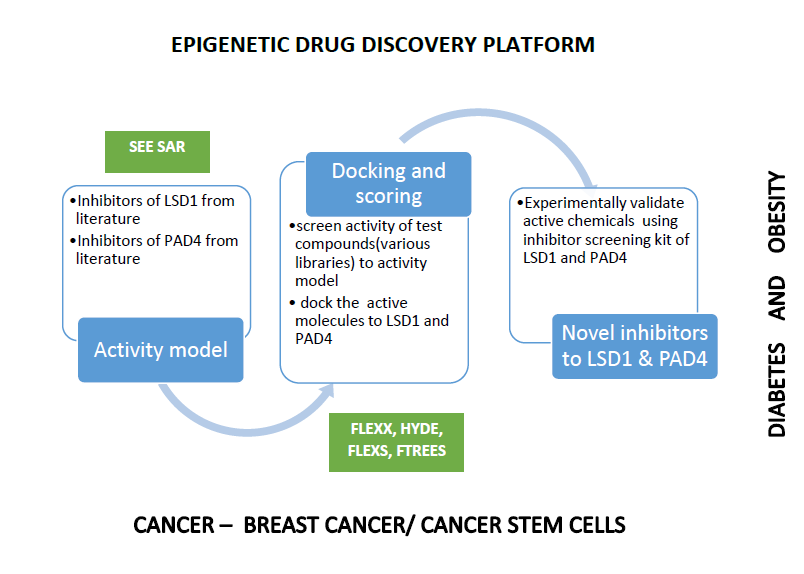
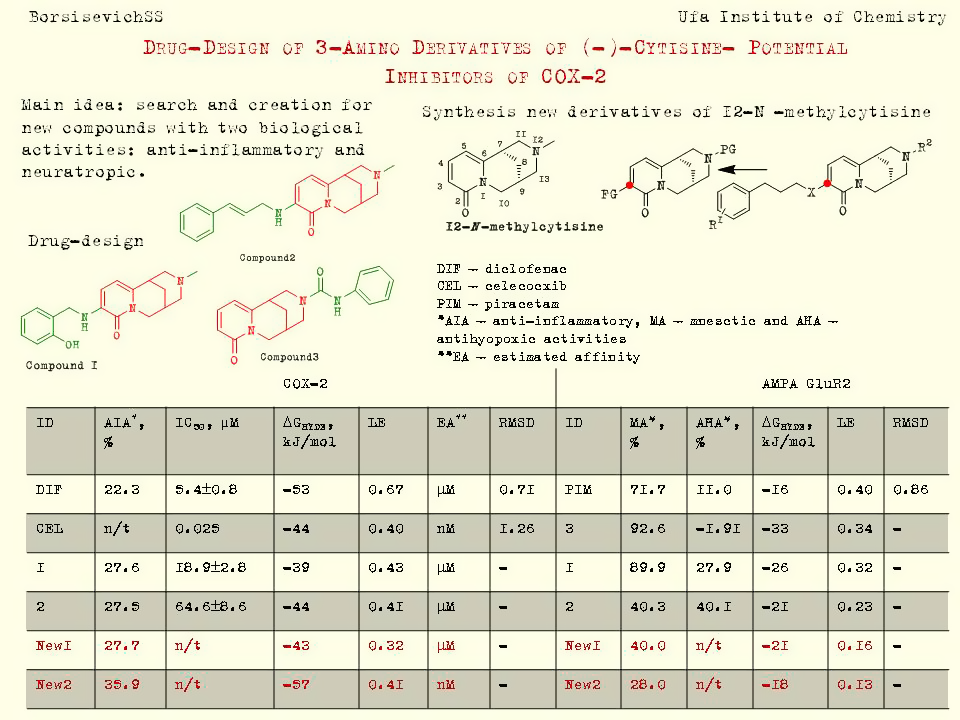
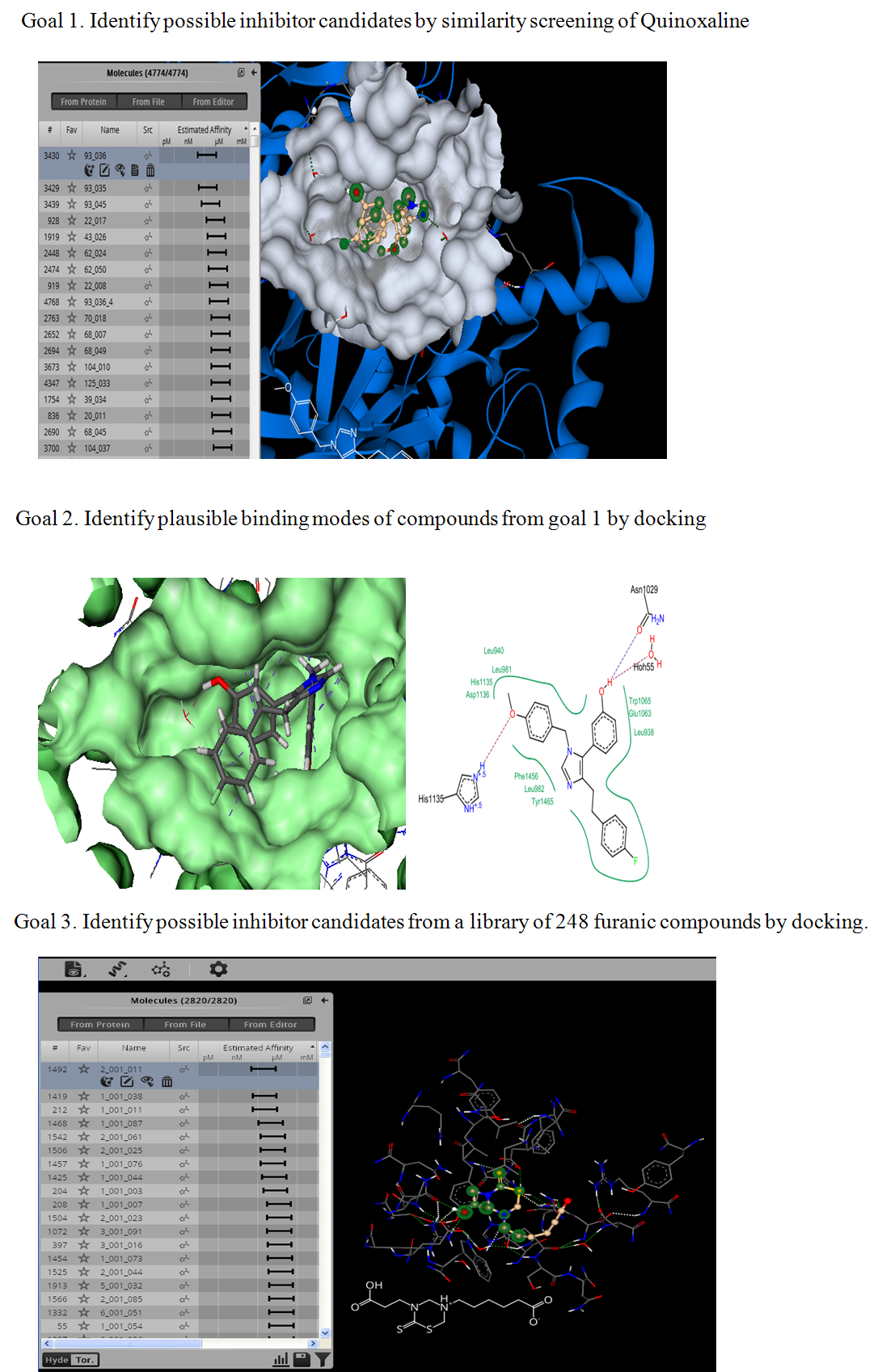
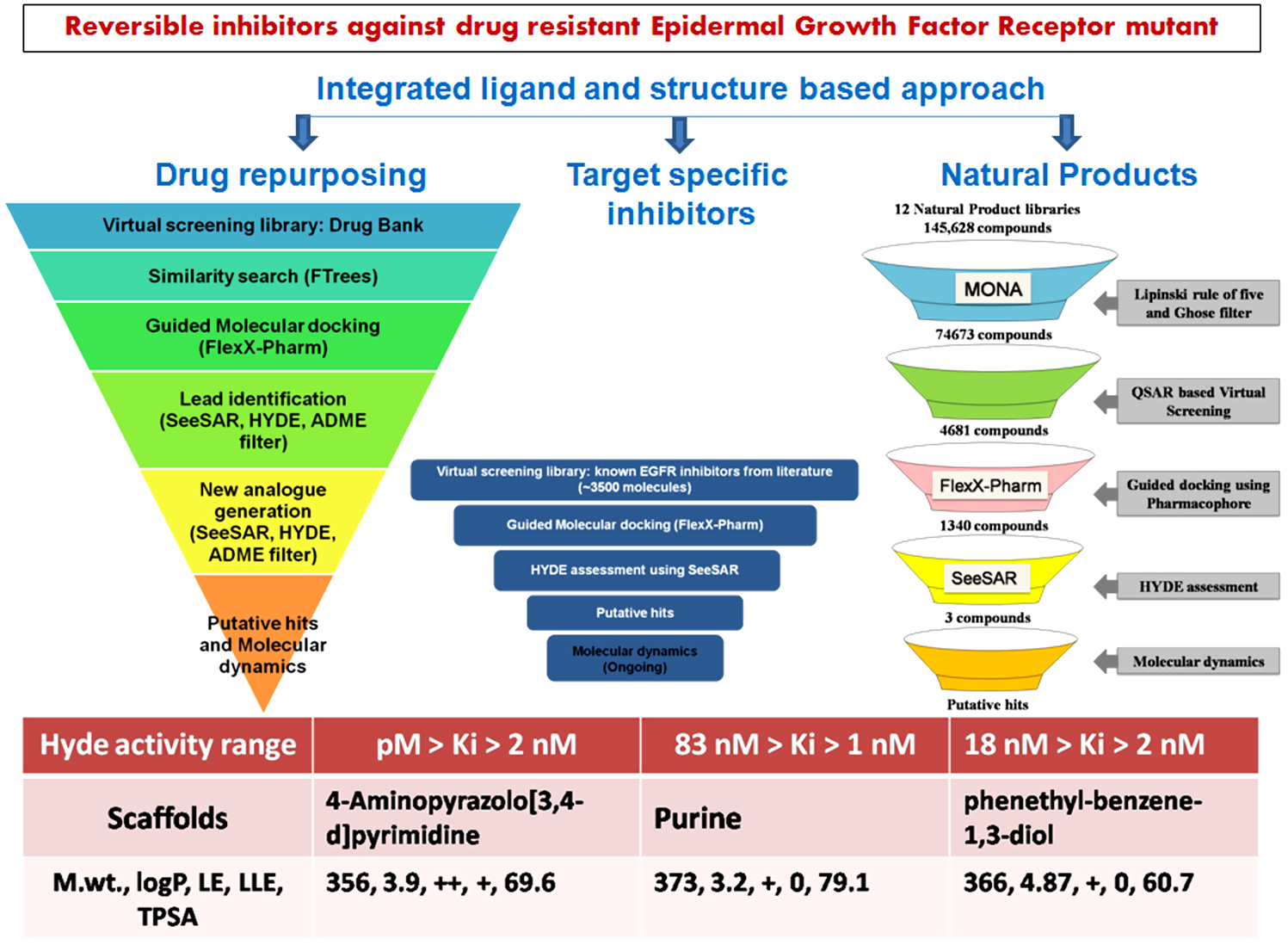
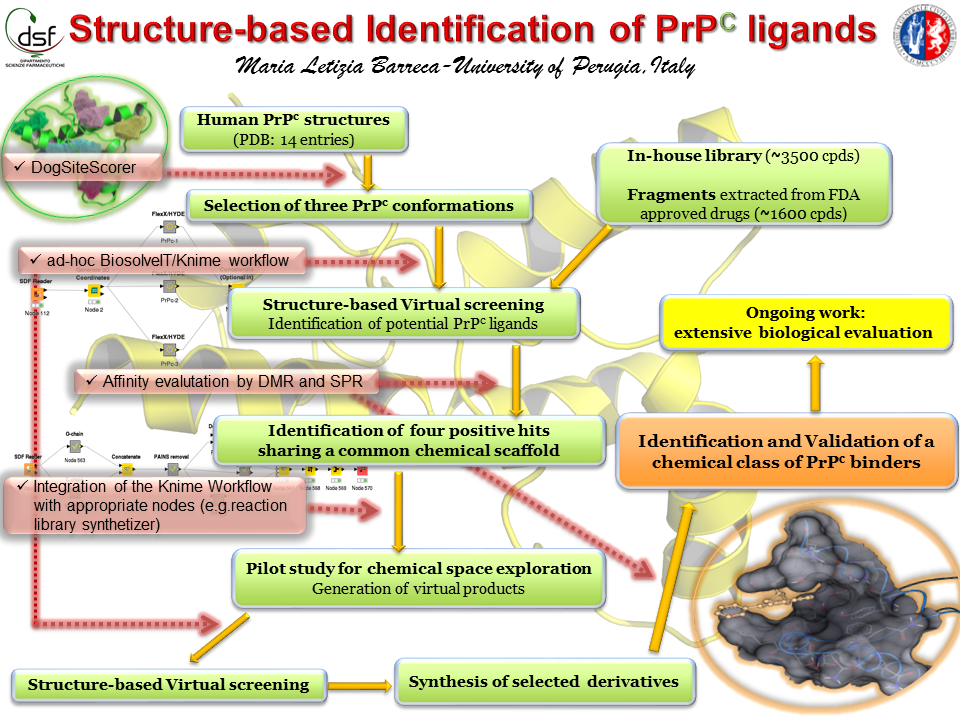
Our project aimed to leverage the BioSolveIT suite of ultra-large chemical space navigation programs to identify make-on-demand agonist candidates for a therapeutically relevant, and structurally unresolved GPCR with few experimentally validated agonists. For this case study, we targeted bitter taste receptor 38 (TAS2R38), a promising drug target for type 2 diabetes given its role as an upstream stimulator of endogenous GLP-1 release. We constructed a homology-based model of TAS2R38 using the closely related TAS2R14 as a template. The homology-based model was refined in its native membrane environment via 20 µs apo molecular dynamics (MD) simulations, and 4 µs of MD...