SeeSAR
Drug Design Dashboard


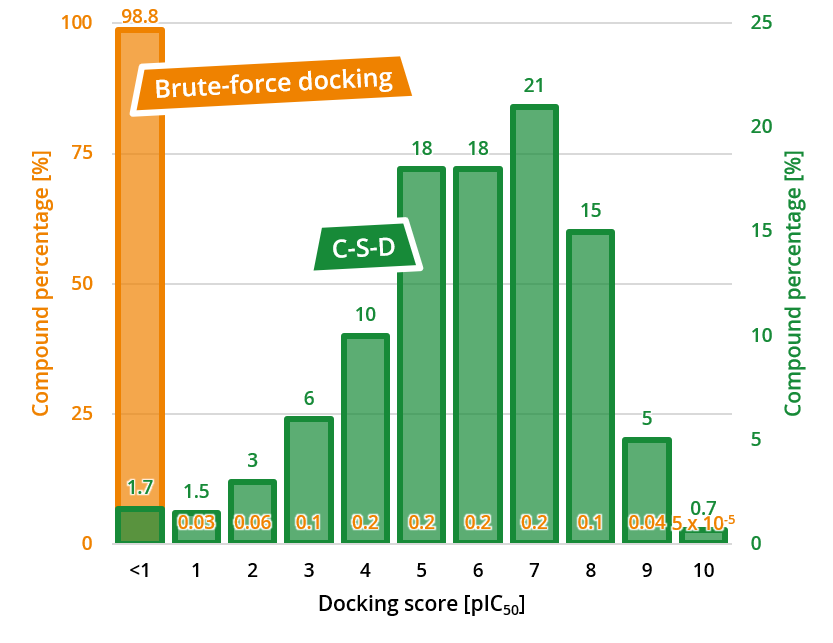
| Category | Requirement | Details |
| HPSee Server — Minimum Hardware Requirements | ||
| Memory (RAM) | 32 GB minimum | Approximately 2 GB per CPU core |
| CPU Cores | 32 cores minimum | Approximately 5–6 days per C-S-D run |
| Disk Space | 500 GB minimum | Allow at least 10 GB per C-S-D project |
| HPSee Server — Operating-System Support | ||
| Linux | Supported | Docker Engine required |
| Windows | Supported | Windows 10 or later, WSL2 and Docker Desktop |
| macOS | Not currently supported | HPSee server installations are not currently supported on macOS |
| SeeSAR Client Requirements | ||
| Memory (RAM) | 16 GB | Required for working with C-S-D results in SeeSAR |
| Operating Systems | Fully supported | Linux, macOS and Windows |
| Download page | Vendor | Size | Order compounds at | |
| Commercial Chemical Spaces [Info] | ||||
| VAST™ | XtalPi | 4.7 × 10⁹ | sales@aifchem.com | |
| AuriVerse | Aurigene | 4.9 × 10⁹ | contactapsl@aurigeneservices.com | |
| GalaXi | WuXi LabNetwork | 2.6 × 10¹⁰ | contact@labnetwork.com | |
| CHEMriya | OTAVAchemicals | 5.5 × 10¹⁰ | info@otava.ca | |
| REAL Space | Enamine Ltd. | 9.5 × 10¹⁰ | libraries@enamine.net | |
| AMBrosia | Ambinter (Greenpharma) | 1.3 × 10¹¹ | ambrosia@greenpharma.com | |
| Freedom Space | Chemspace | 3.0 × 10¹¹ | sales@chem-space.com | |
| xREAL Space | Enamine | 4.4 × 10¹² | libraries@enamine.net | |
| eXplore | eMolecules | 8.3 × 10¹² | explore@emolecules.com | |
| Synple Space | Synple Chem | 8.3 × 10¹² | order@synplechem.com | |
| Virtual Chemical Spaces | ||||
| KnowledgeSpace | BioSolveIT (virtual) | 2.6 × 10¹⁴ | ||
| SAVI Space | BioSolveIT (virtual), Enamine* | 7.5 × 109 | libraries@enamine.net* | |







