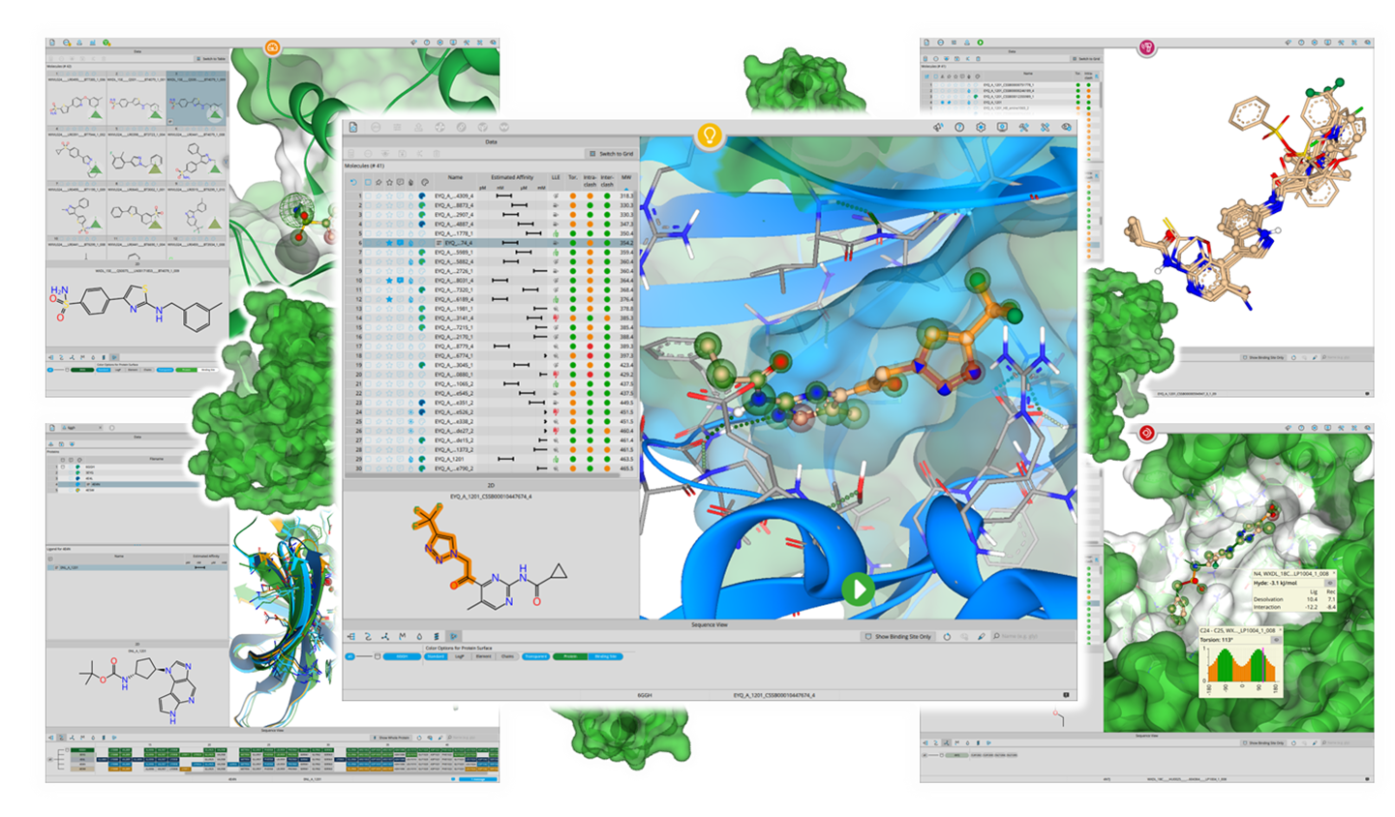
SeeSAR
Drug Design Dashboard
 SeeSAR
SeeSAR











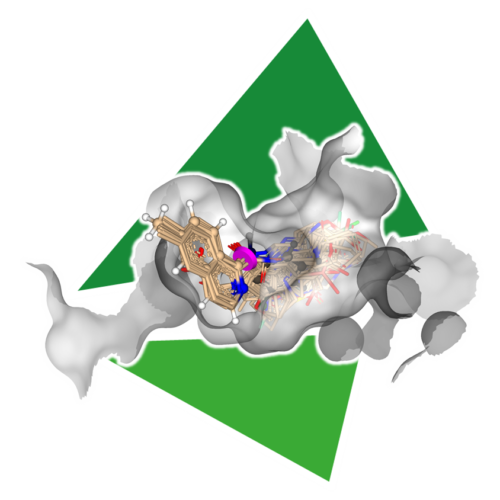
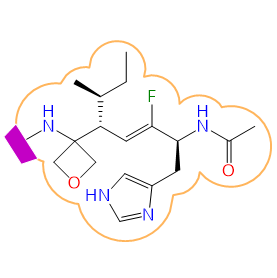
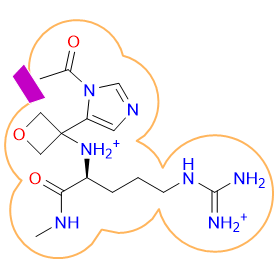
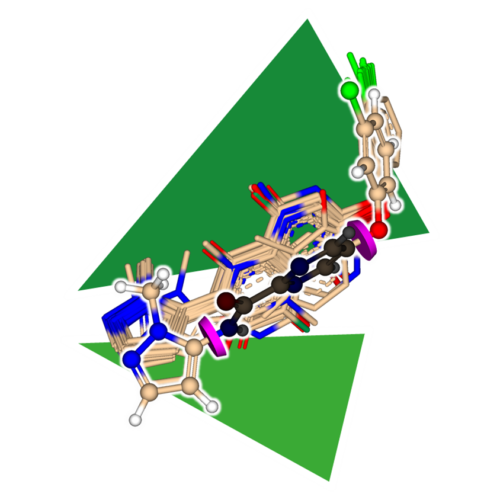

For older versions and an elaborated list please visit here.

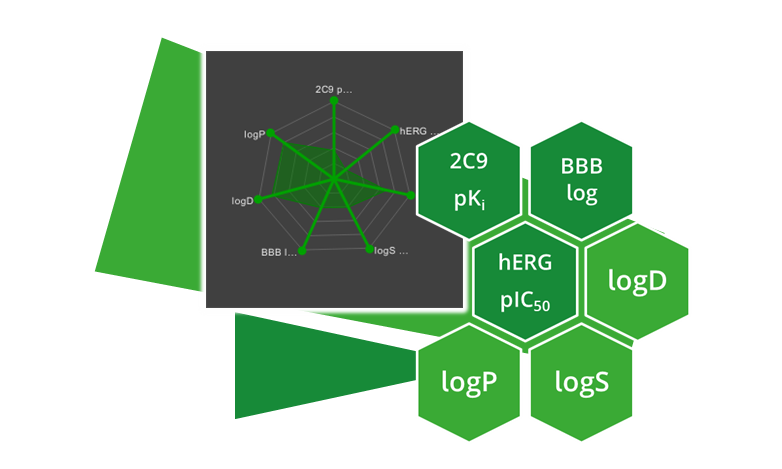
| ADME parameter | |
| 2C9 pKi | Cytochrome P450 CYP2C9 pKi prediction. Affinity prediction of the compound to bind at the enzyme involved in several metabolic drug pathways. |
| 2D6 affinity category | Cytochrome P450 CYP2D6 classification. Compounds are predicted to be in one of four categories: ‘low’ for compounds with a pKi<5, ‘medium for compounds with a pKi between 5 and 6, ‘high for compounds with a pKi between 6 and 7, and ‘very high’ for compounds with a pKi>7. |
| BBB category | Classification of compounds into ‘+’-category if log([brain]:[blood])≥-0.5 and ‘-’category if log([brain]:[blood])<-0.5. |
| BBB log([brain]:[blood]) | Logarithm of brain-blood partition coefficient of a compound. Can be used as indicator for CNS active compounds. |
| HIA category | Human intestinal absorption classification. Compound which are predicted to be absorbed ≥30% are classified with ‘+’, compounds which are predicted to be absorbed <30% are classified with ‘-’. |
| P-gp category | P-glycoprotein 1 (also known as multidrug resistance protein 1 (MDR1) and ATP-binding cassette sub-family B member 1 (ABCB1)) transport classification. Predicts if a compound is a substrate of P-gp. |
| PPB90 category | Plasma protein binding classification. Predicts a classification of ‘low’ for compounds which are <90% bound and high for compounds which are >90% bound. |
| hERG pIC50 | Prediction of pIC50 values for inhibition of human Ether-a-go-go Related Gene (hERG) potassium channels expressed in mammalian cells. |
| logD | Logarithm of n-octanol-water partition coefficient (also known as n-octanol-water partition ratio) at fixed physiological pH 7.4. Used to describe the relationship between lipophilicity and hydrophilicity of an ionized compound. |
| logP | Logarithm of n-octanol-water partition coefficient. Used to describe the relationship between lipophilicity and hydrophilicity of a neutral compound. |
| logS | Logarithm of intrinsic aqueous solubility in µM for neutral compounds. |
| logS @ pH 7.4 | Logarithm of intrinsic aqueous solubility at physiological pH 7.4 in µM for ionized compounds. |