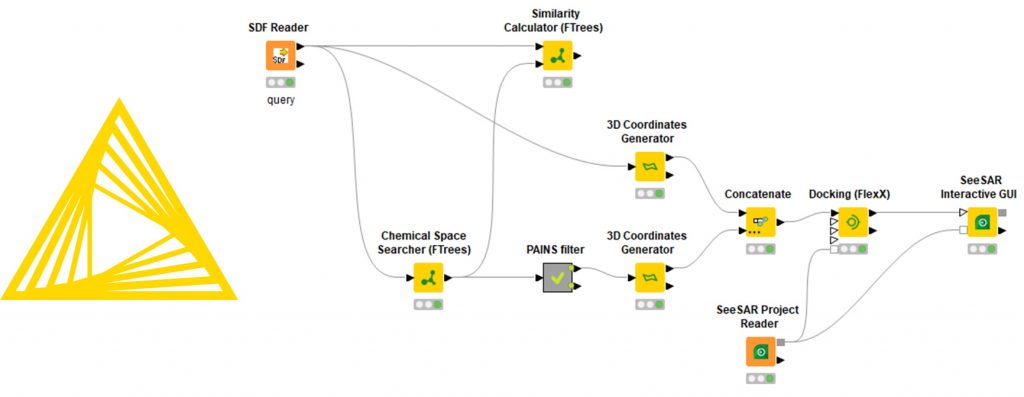
Similarity searching is a task that needs to be performed routinely and oftentimes in conjunction with other tools in a workflow. The FTrees Similarity node compares the molecules in the pipeline to separately provided query molecules. Additionally you may:
- filter molecules according to similarity
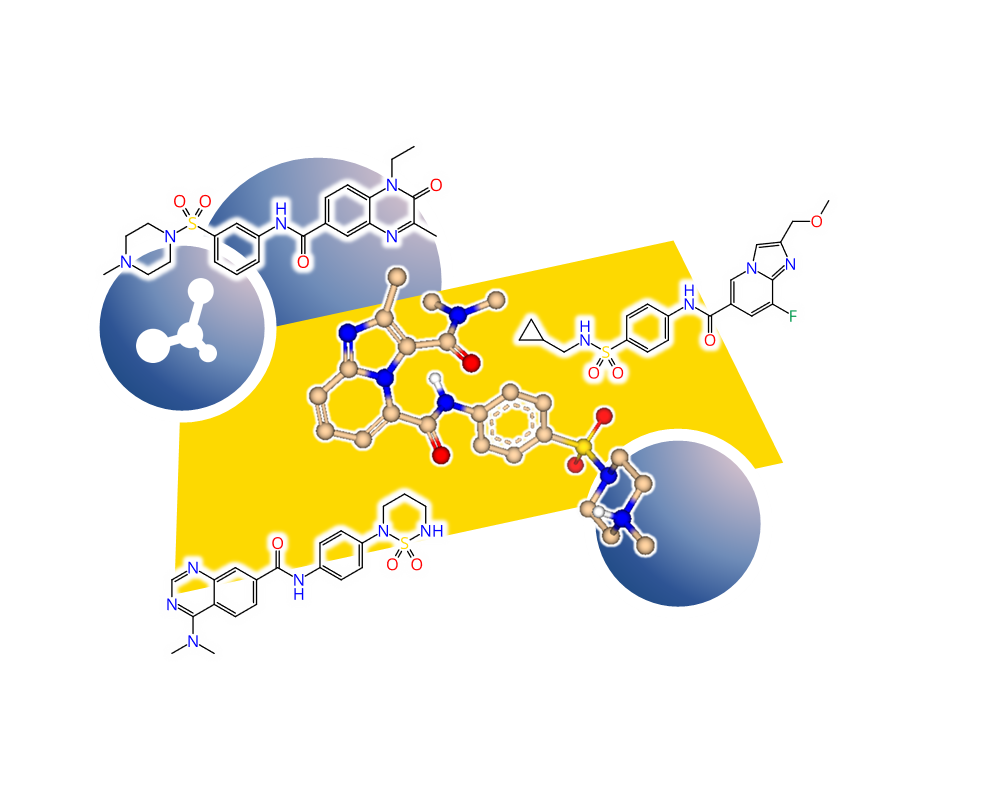
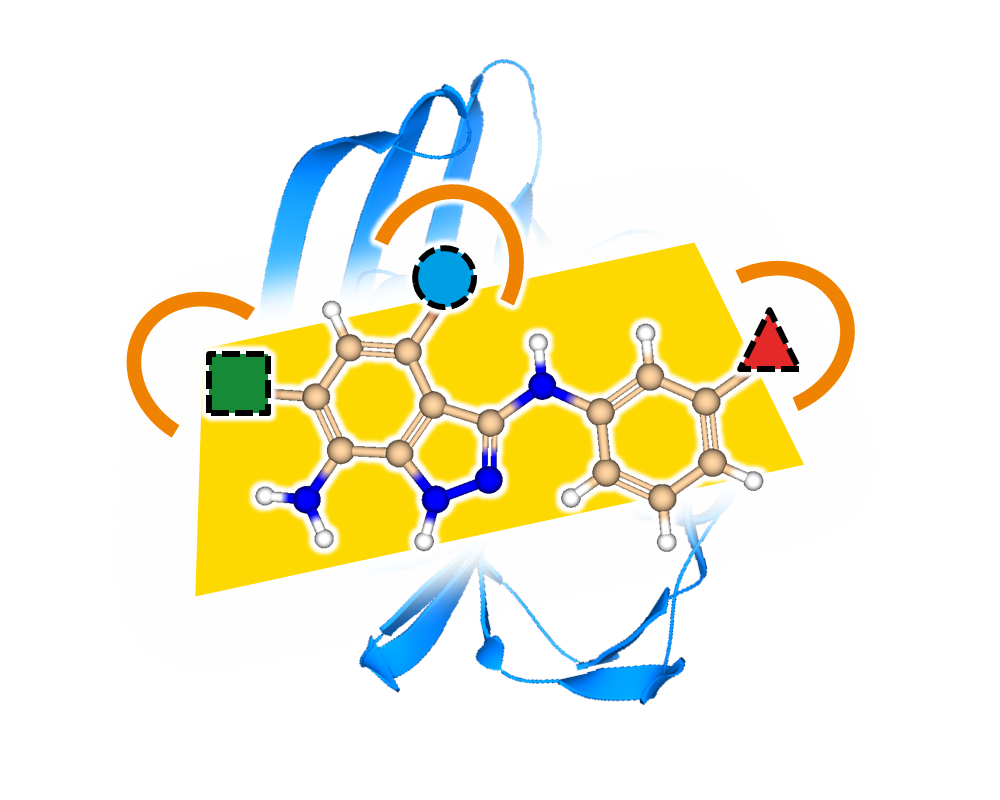
- generate 2D depictions that visualize the FTrees-mapping using colors
- compute 3D alignments based on the FTrees-mapping on the fly
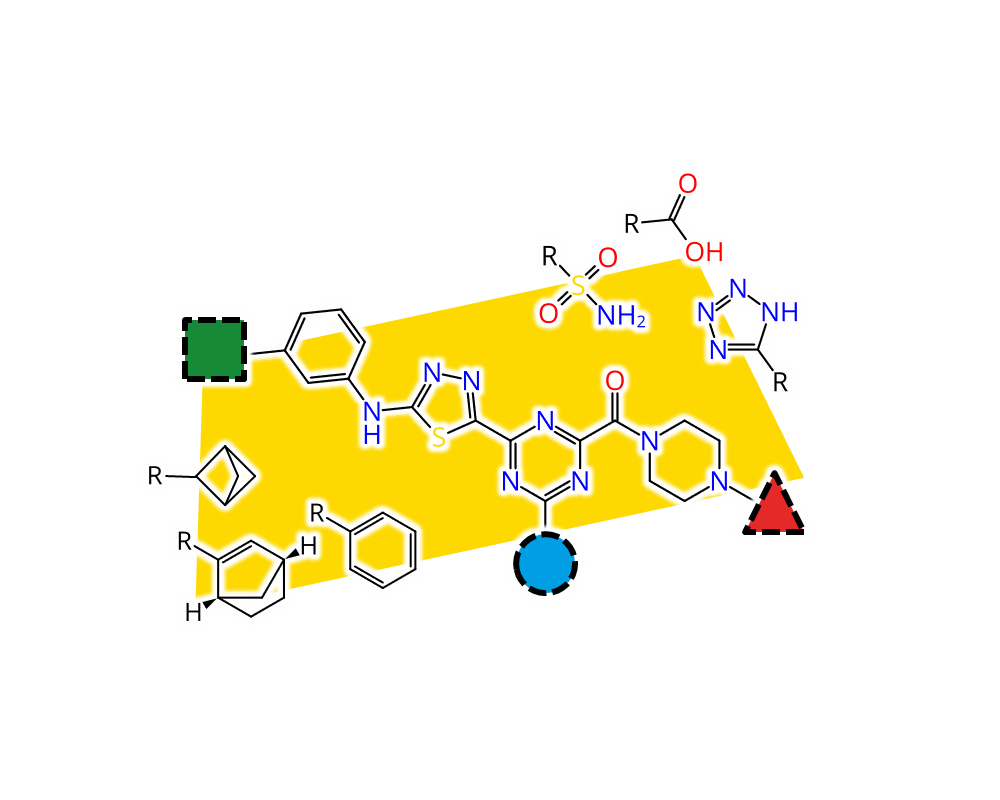
Feature Trees Fragment Spaces (FTrees-FS) is a unique technology, which enables the user to perform similarity searches in innumerably large compound spaces. The KNIME
®-module comprises the search engine. You may use it for example to perform similarity searches in over 12 billion molecules of the so-called KnowledgeSpace
™, which based on chemistry protocols to ensure it comprises synthesizable molecules. The KnowledgeSpace
™ can be downloaded free of charge
here.
 KNIME
KNIME