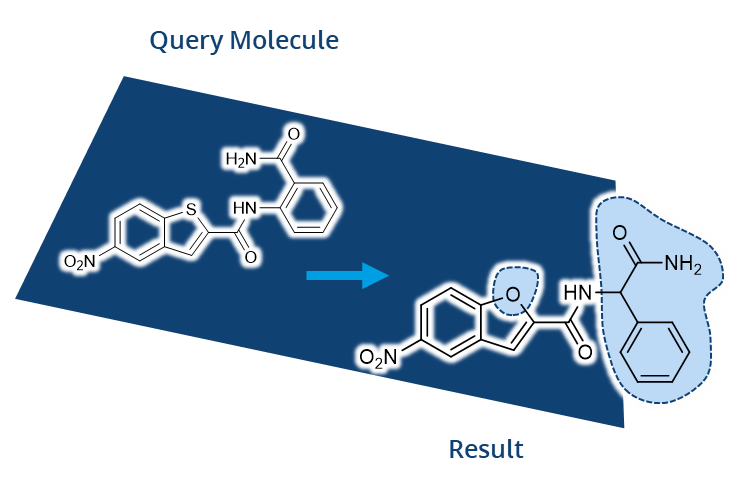
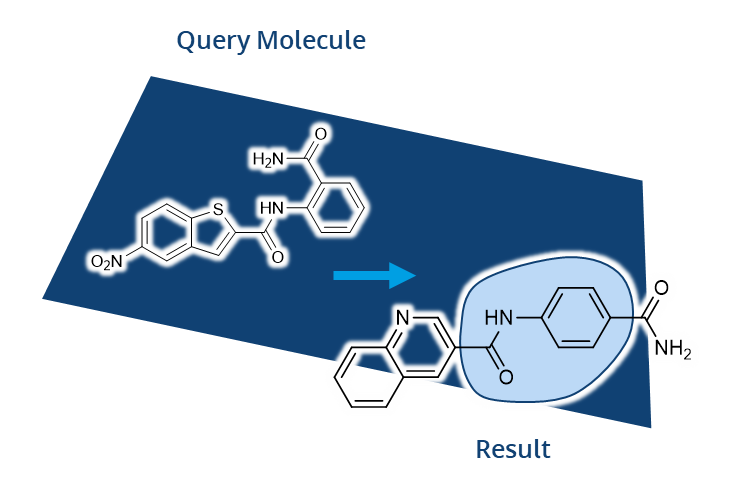
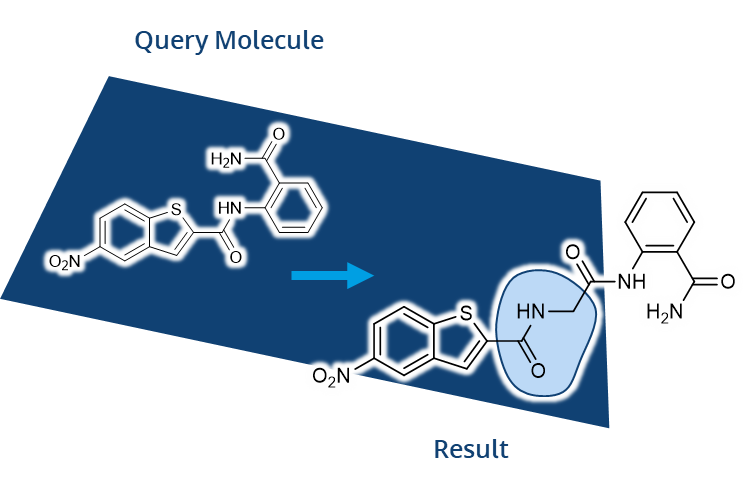
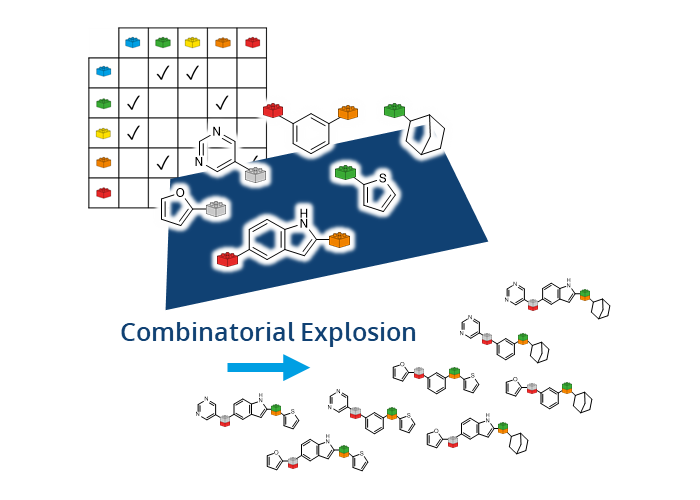
FTrees by Matthias Rarey was among the first tools developed that allowed users to later search for compounds in Chemical Spaces with beyond-billions of molecules. In FTrees, query molecules are translated into fuzzy pharmacophore descriptors. Subsequently those are used to search at unprecended speed for similar molecules in the Chemical Space with a tree alignment approach. Finally, users receive results with numerical descriptors to understand the similarity.
FTrees is part of the Chemical Space navigation platform
infiniSee and can be used as standalone commandline tool for programmatic access to the technology. Further information can be found
here.