The discovery process of suitable drug candidates requires a reliable Swiss Army knife software toolkit with comprehensible results. Interplay between smooth operation, visualized structure-activity relationships, transparent science, interactive feedback, and calculation speed are key to successful projects. SeeSAR is every modelers SAR and
ADME dashboard, created to support and lead every drug development process. With the SeeSAR platform you can estimate affinities with
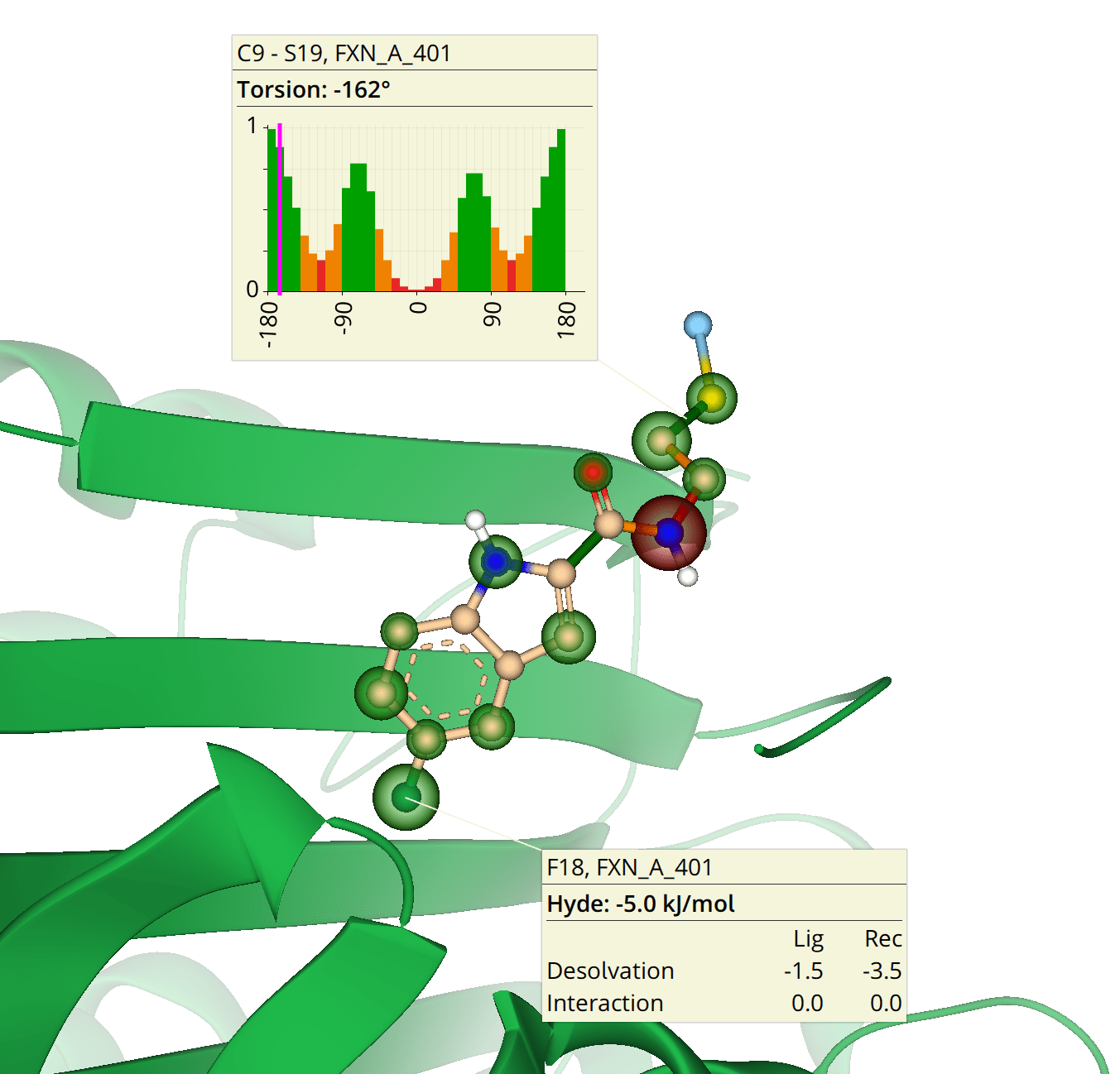
HYDE binding assessment and phsyicochemical properties of your compounds within seconds. Explore binding sites and perform docking studies with you ligands. With our provided indexed fragment libraries you can link molecule fragments, grow into unoccupied binding cavities, or even replace the very core of your compound with a suitable scaffold. The visual 3D guidance supports you in your decision making-process and inspires you with creative solutions and structures. Experience drug discovery like never before.