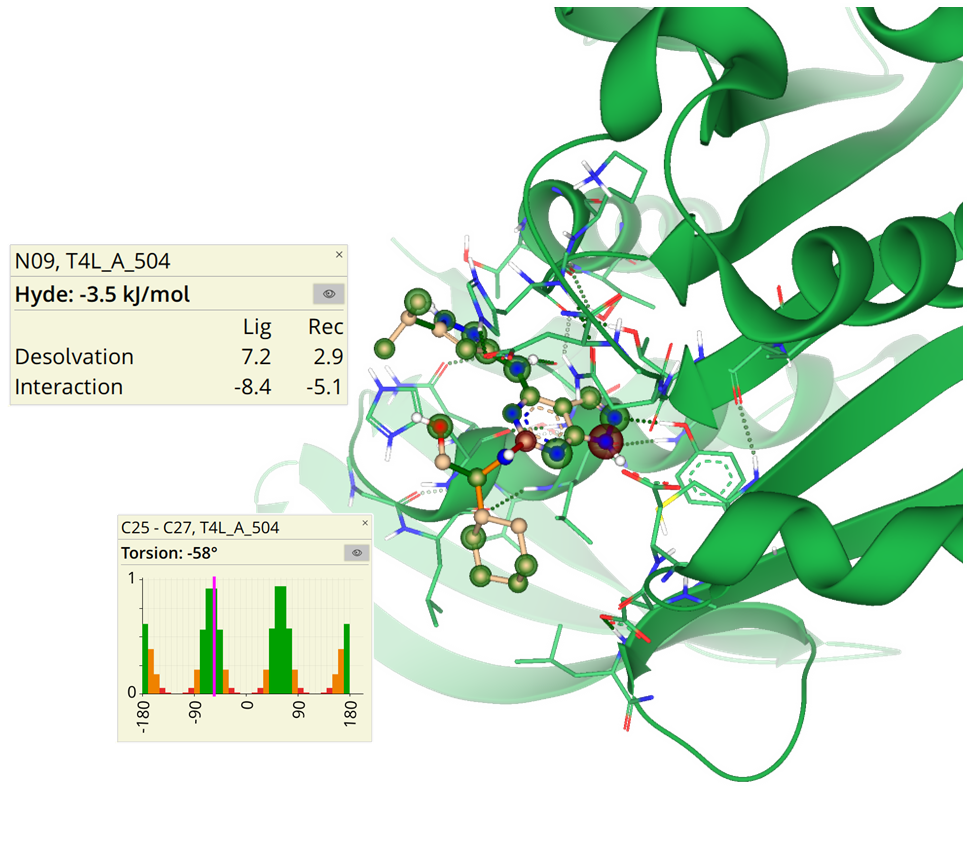
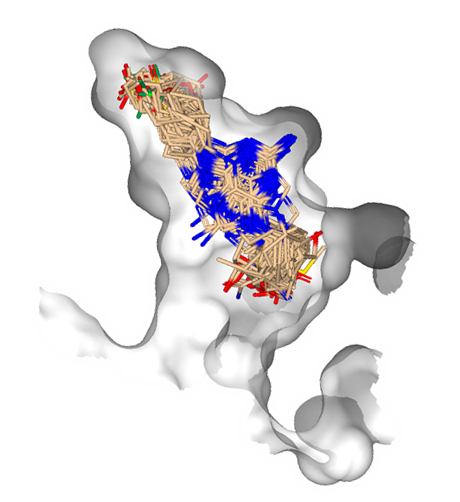
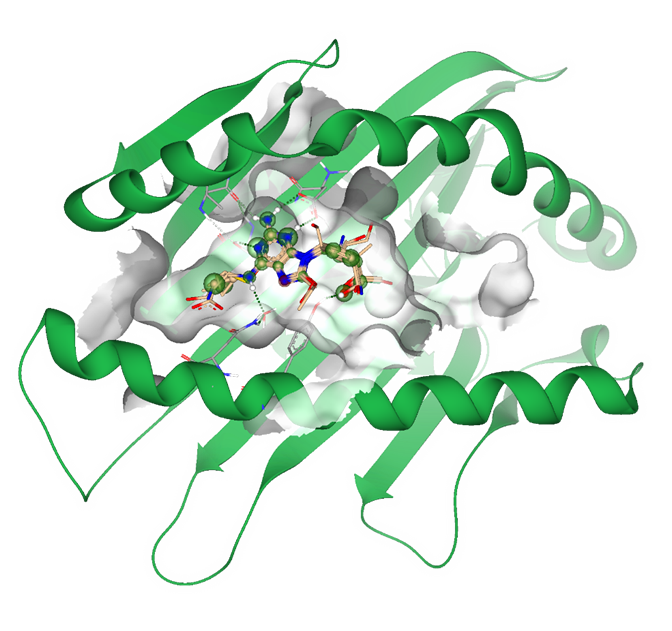
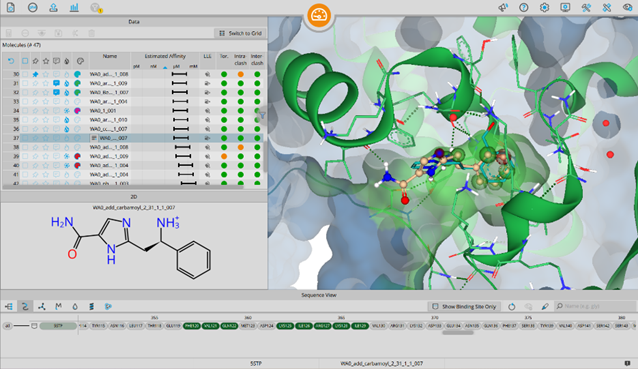
SeeSAR can be applied in different areas of research: real drug targets, agrochemistry, food, cosmetics, microbiology, and many more.
Applying industry-relevant methods users can propose modifications to a ligand, generate new compound ideas, and apply principles used in modern hit identification and lead optimization. SeeSAR covers the entire spectrum of molecular modeling, making it versatile and applicable across disciplines.
- Users who typically work in the lab quickly find their way around the software.
- The low entry barrier allows, for example, synthetic chemists to come up with their own ideas — with just a few clicks — for which compound to synthesize next to improve activity or to identify where there is room to expand the molecule.
- Easy access to the world of molecular modeling adds a valuable skill to the user's portfolio, enhancing interdisciplinary understanding.