Introduction to Docking
This introduction to docking will help molecular modelling beginners to understand the basics of binding mode prediction, scoring, and virtual screening.
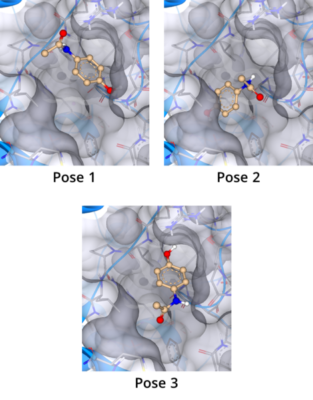
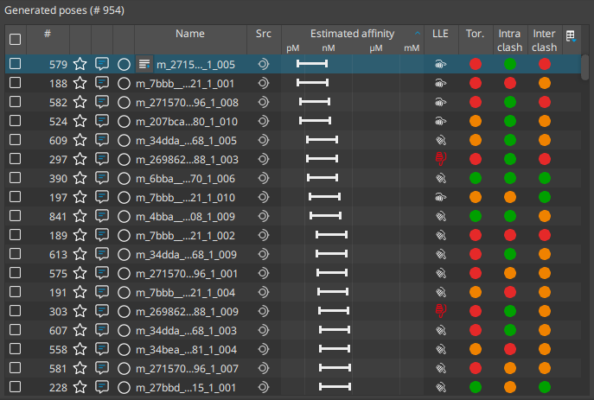
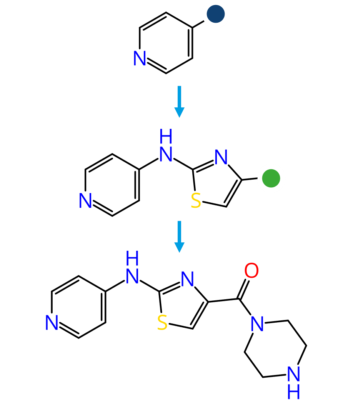
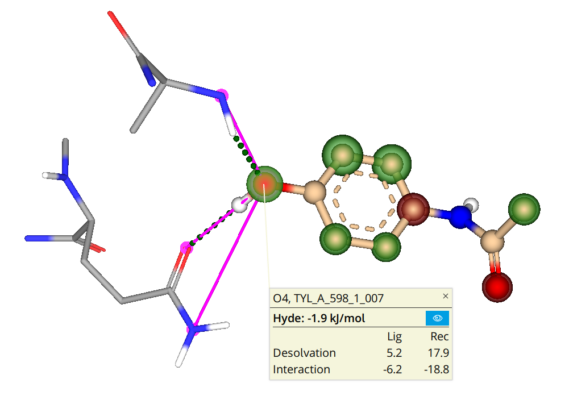
Molecular docking is a computational approach that belongs to the field of molecular modelling. It describes the prediction of the orientation of a molecule at a target structure (e.g. protein, RNA, DNA). Knowledge about the arrangement of a molecule in a binding site is the perquisite to assess the quality of the interactions it may form with the structure at atomic level.