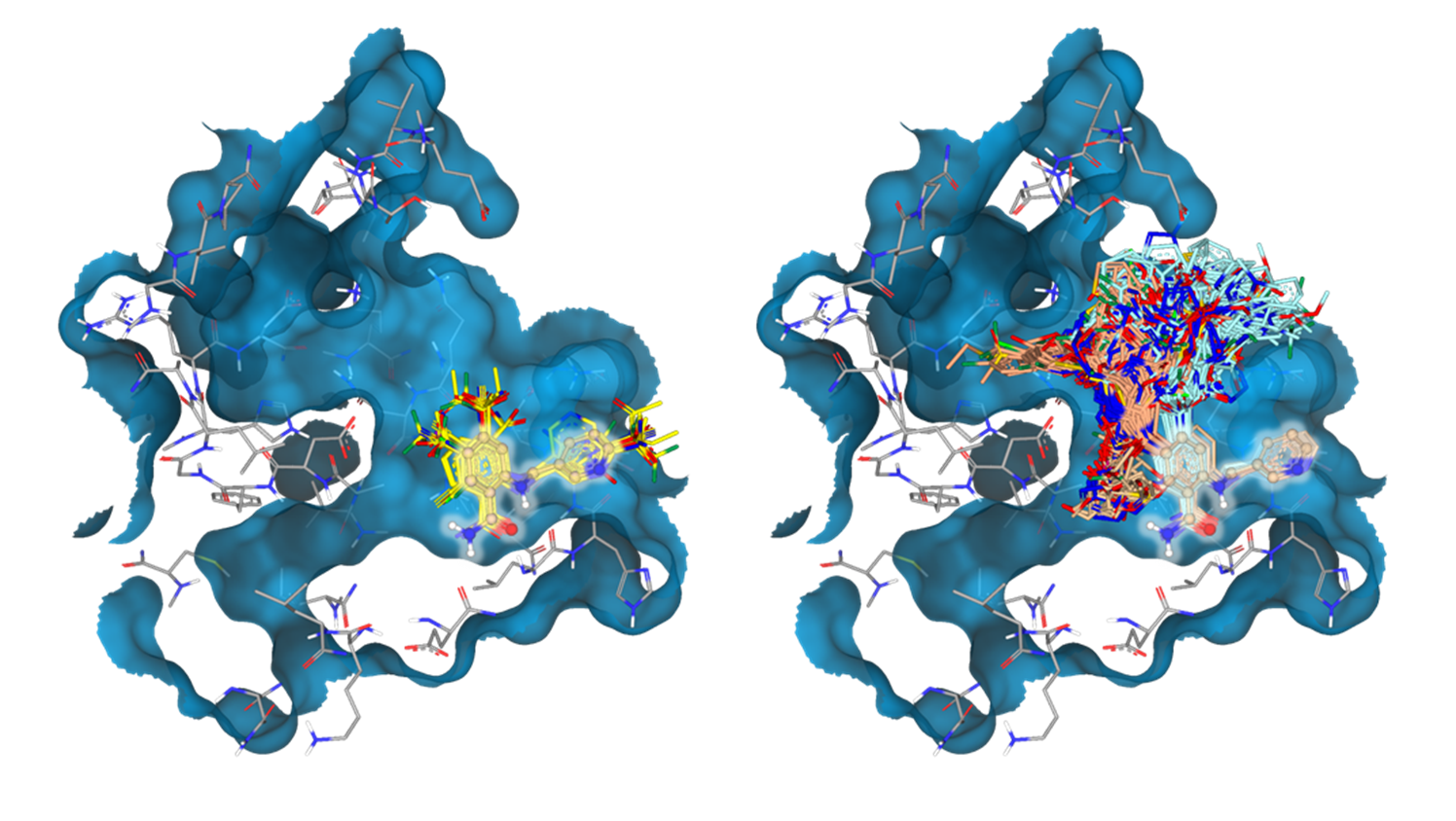
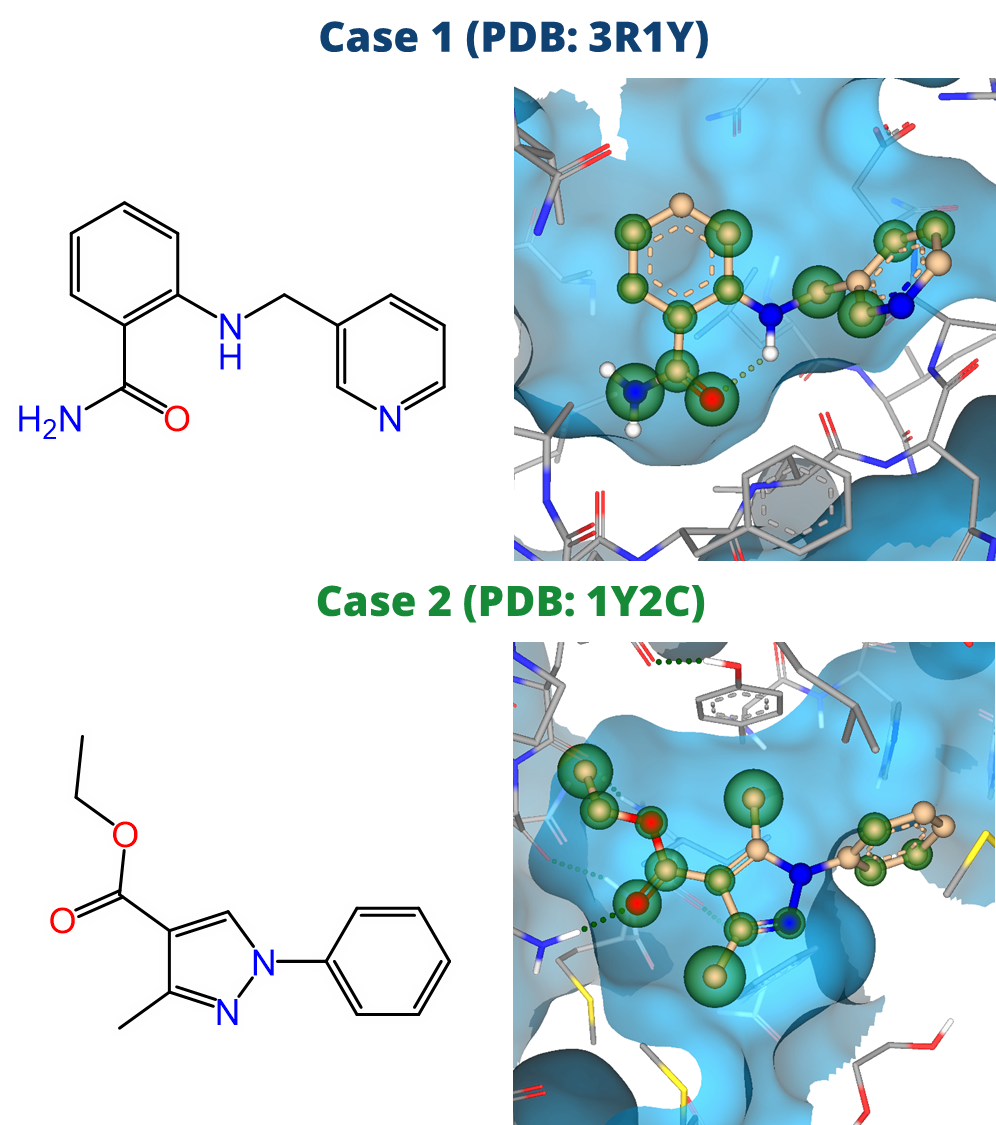
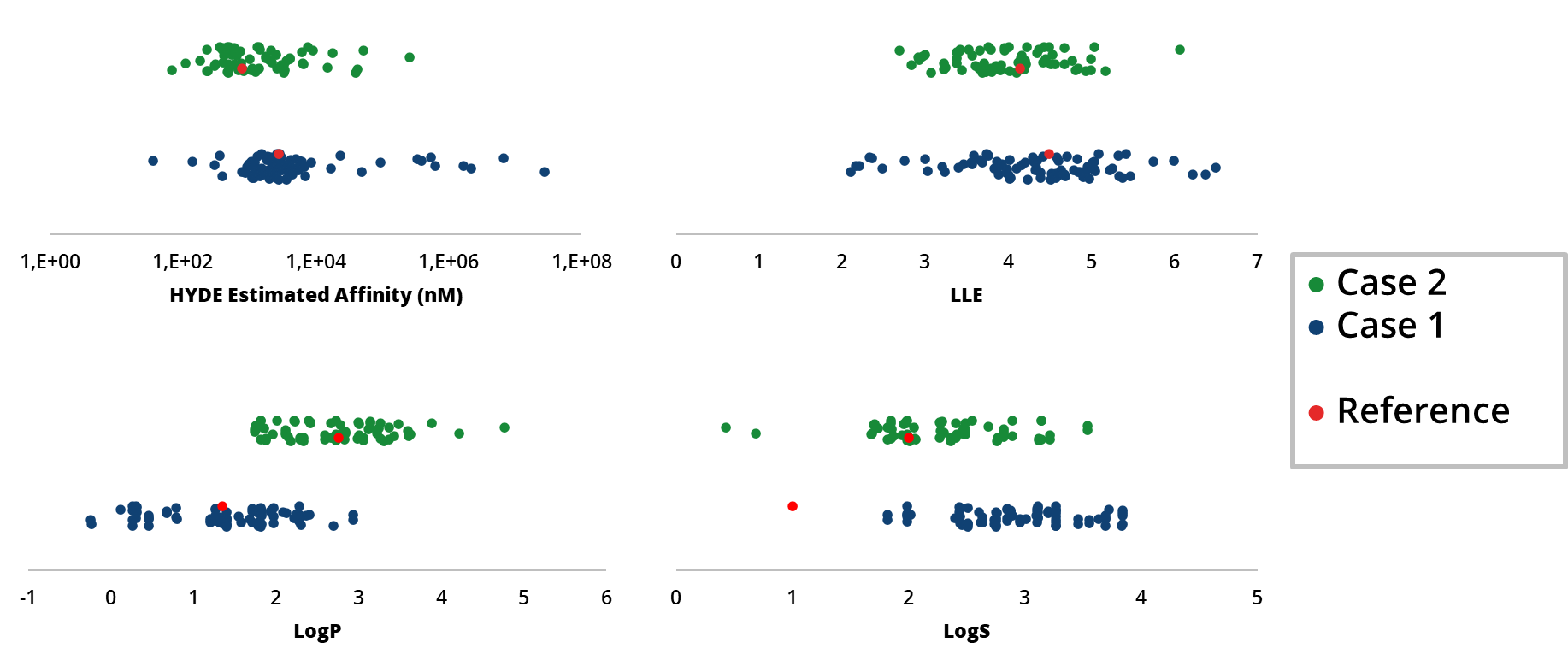
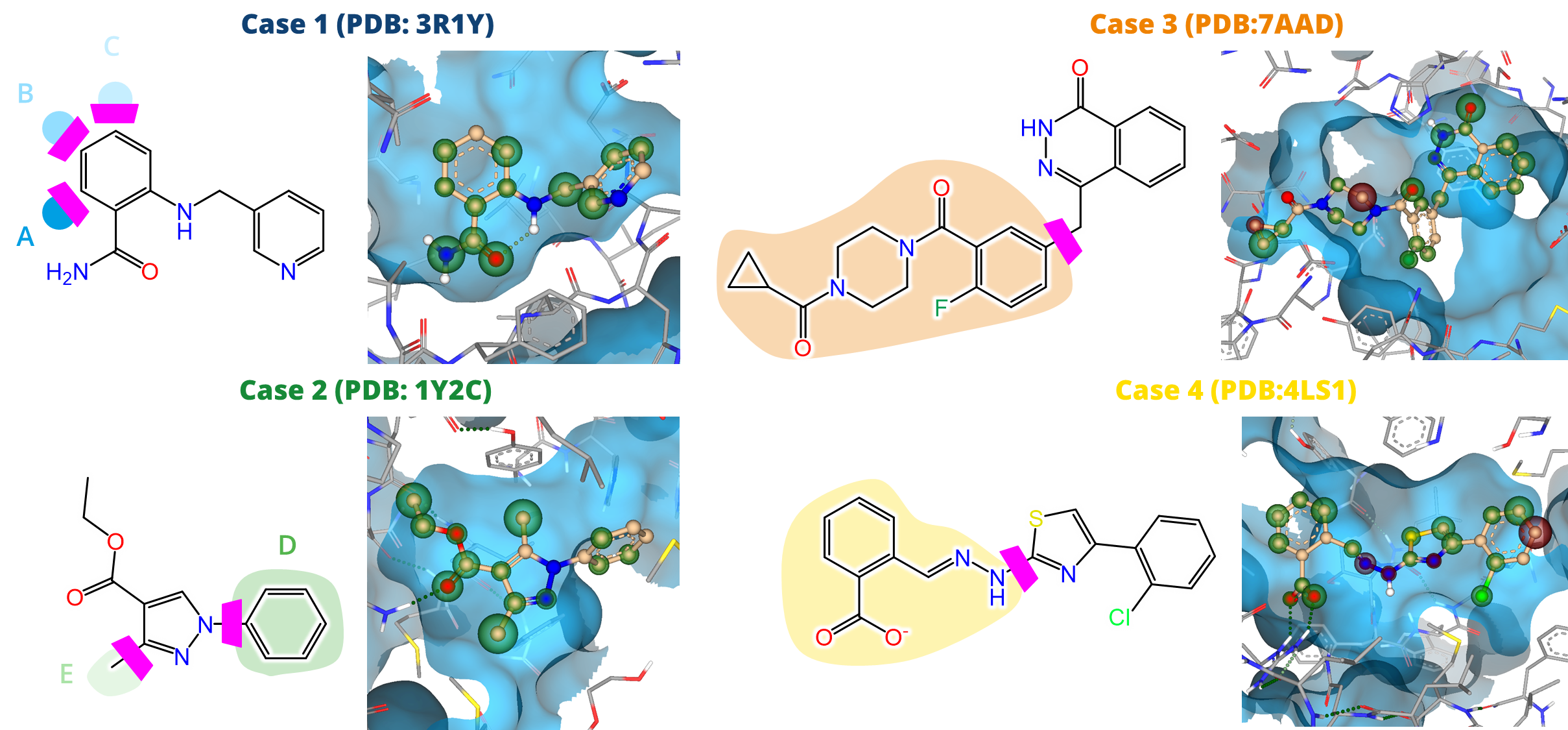
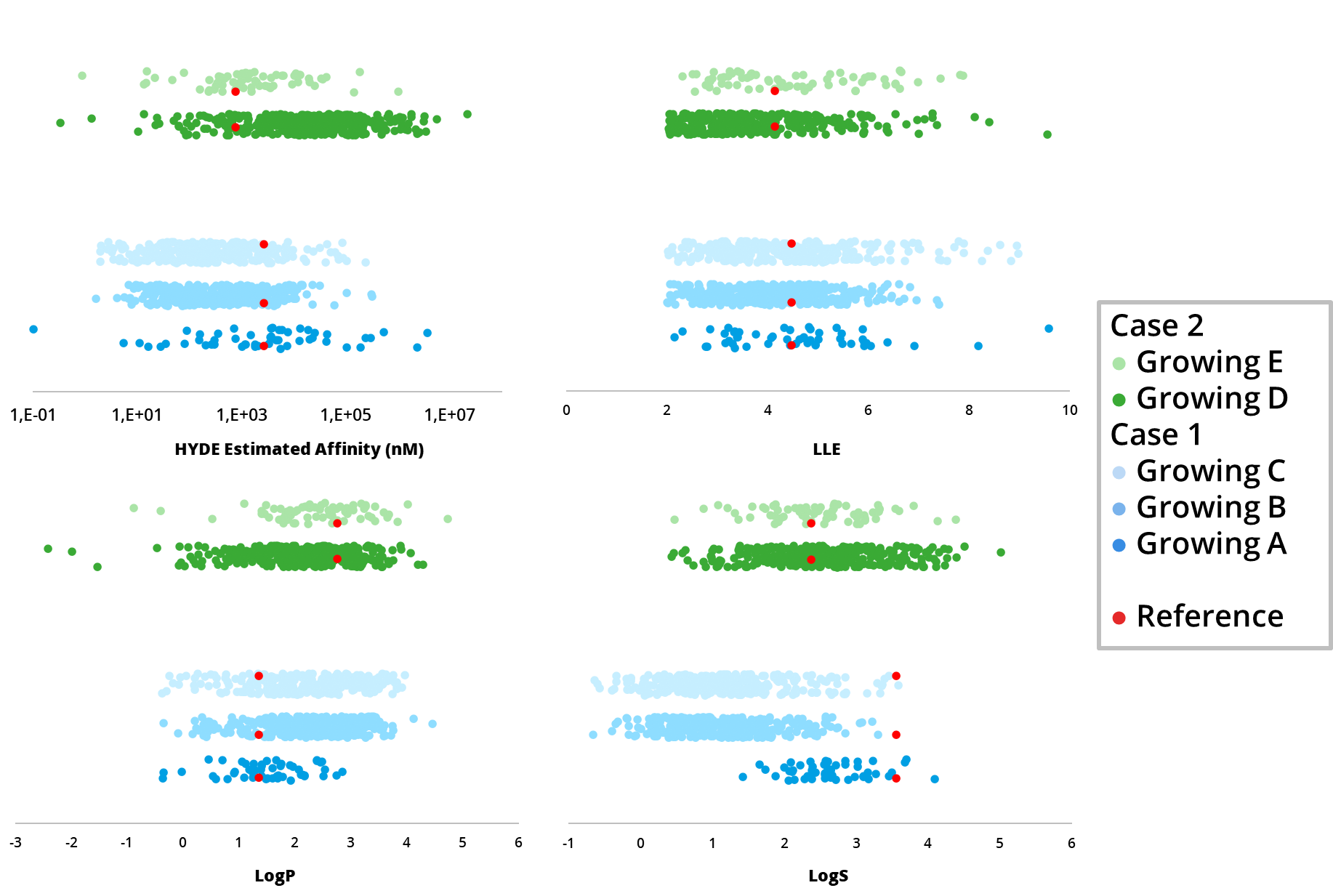
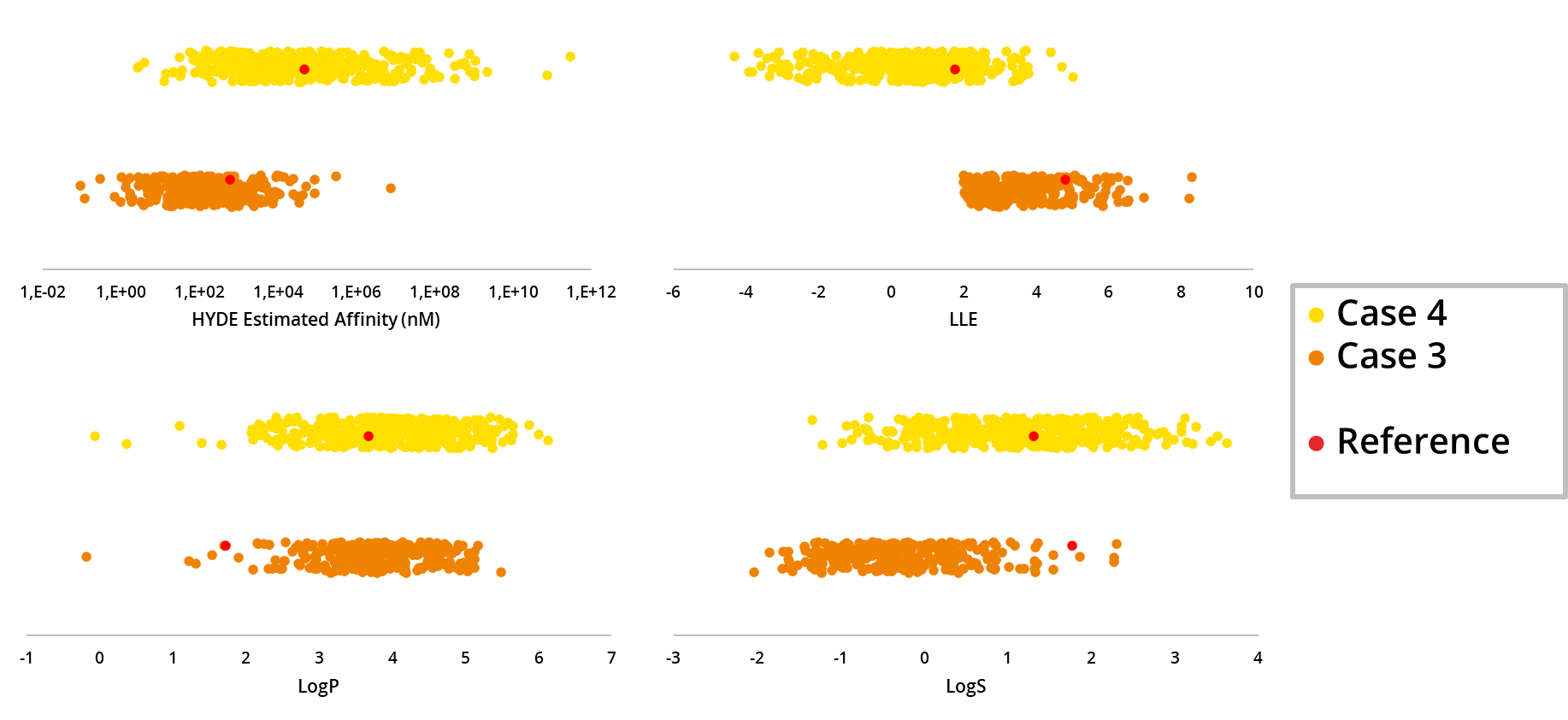
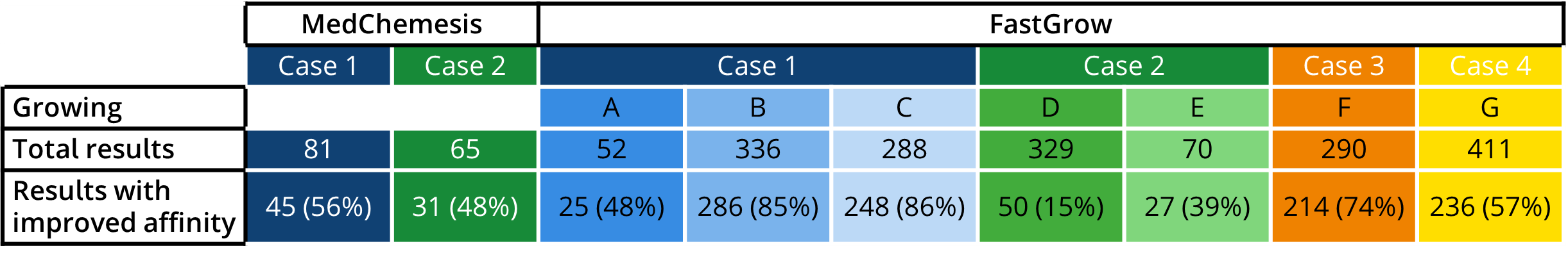
In the initial stages of early drug discovery, the focus is often on compound ideation: Investigating the Chemical Space around a compound of interest via structure-guided screening for decorations and modifications.
This includes the assessment of possible modifications to improve the physicochemical properties of the drug candidate and/or the addition of decorations forming high-quality interactions with the target to improve the potency of the compound.
In this application spotlight, we used the tools
MedChemesis and
FastGrow, both part of our
drug design dashboard SeeSAR, on several drug discovery scenarios. We highlight their potential as valuable resources for ideation of improved drug candidates in the context of predicted potency, ligand-lipophilicity efficiency (LLE), and important ADME properties such as logP and logS.