KNIME Support
Unfortunately, KNIME is no longer actively supported by BioSolveIT.
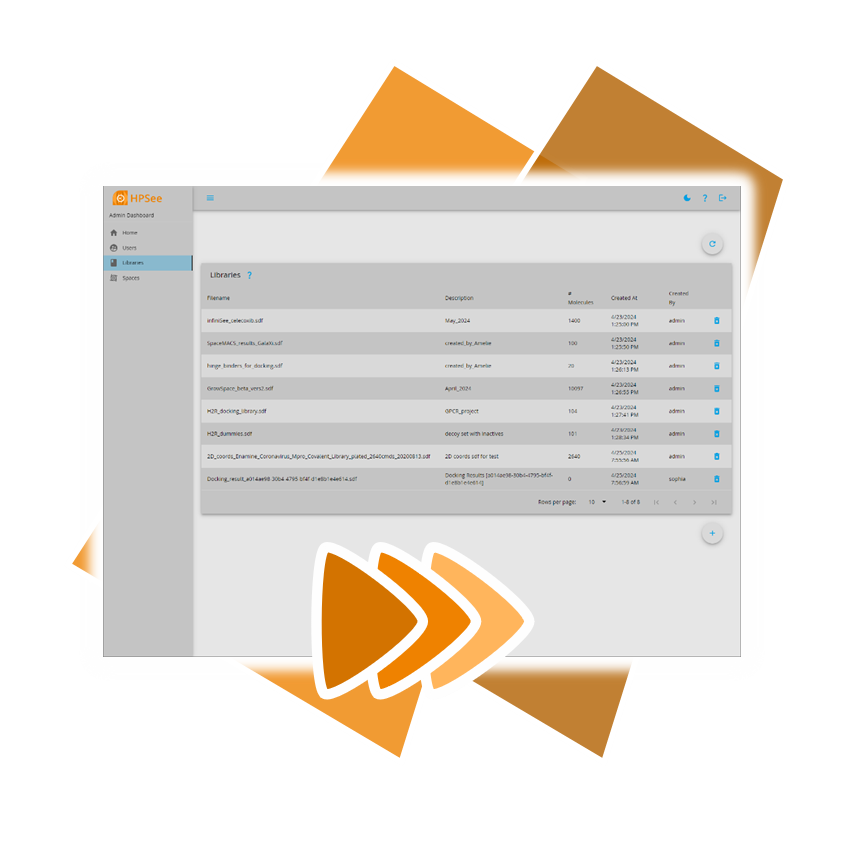
However, there's no need to rely on outdated solutions, as BioSolveIT introduces a new workflow environment tailored to the needs of computational chemists and data scientists: HPSee.