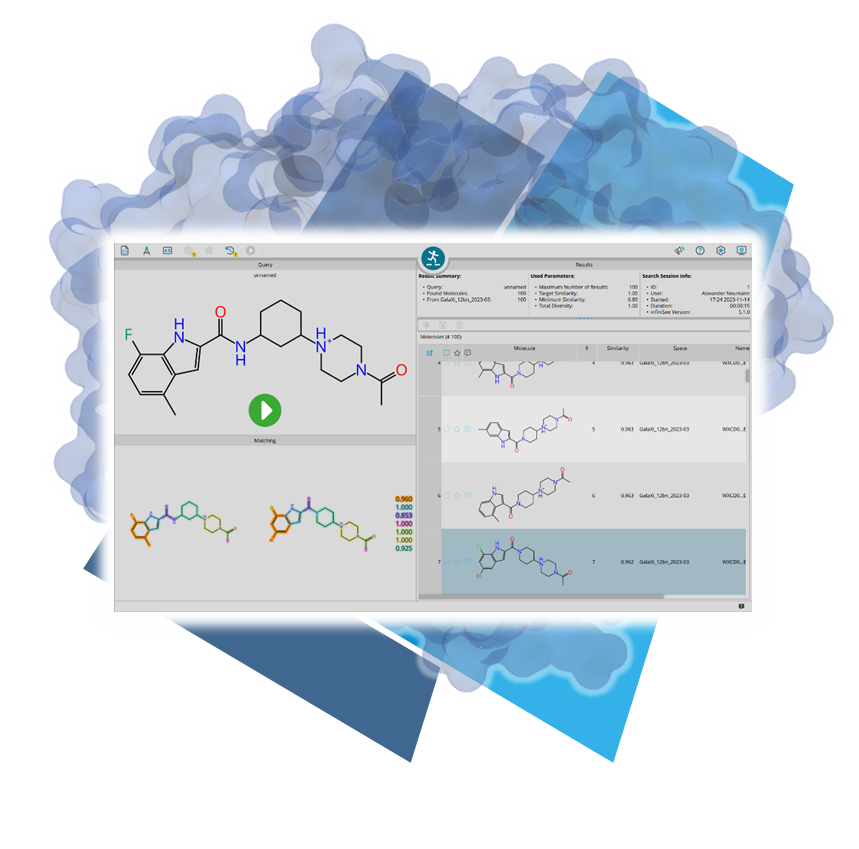
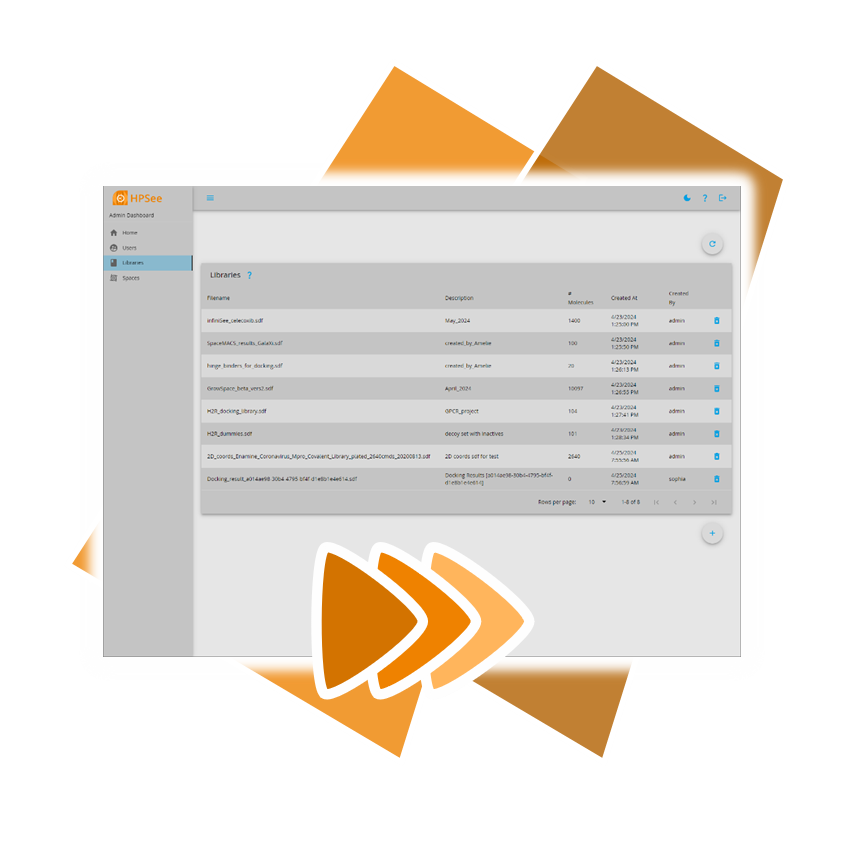
BioSolveIT Platforms
Our trusted platforms were developed and tailored to address the needs of modern drug discovery challenges. Equipped with an advanced graphical user interface, they can be intuitively operated by both novices and experienced professionals, fostering an immersive experience that paves the way for success.
Usingour published algorithms and tools, referred to as 'components,' both platforms — the drug design dashboard SeeSAR and the Chemical Space navigation platform infiniSee — employ them as their engine to promptly deliver dependable and innovative results.