


SeeSAR’s Activity Spotter Mode is designed to dismantle the barrier of raw data and actionable SAR.
It helps to answer the most relevant fields in hit-to-lead and lead optimization campaigns: 3D SAR and pharmacophore modeling. Which structural features in my molecule set are associated with activity? Which ones are linked to inactivity?
In the known BioSolveIT fashion, Activity Spotter addresses this process fast, visual, and highly practical for real project work of medicinal and computational Chemists. It is built for scientists who want more than generic pattern recognition. Deriving tangible 3D hypothesis from aligned compounds, it guides design and screening campaigns with rational insights and comprehensive relations.
Key Aspects
- Integrated Binary Logic: It treats the design process as a series of “preferred” vs. “non-preferred” outcomes.
- Multidimensional Utility: Beyond activity, it can be used to map selectivity, toxicity, metabolic stability, or covalent reactivity.
- Diverse Chemotypes: Unlike tools limited to rigid series, Activity Spotter can derive features from closely related analogs AND diverse chemotypes simultaneously.
Did you know?
SeeSAR offers an extensive selection of possible pharmacophore constraints that can be placed afterwards. Beyond common definitions such as H-bond acceptors and donors, it is also possible to define interaction partners on the ligand itself, starting from the target, as well as to choose more exotic definitions such as spiro centers or bicycles.
In addition, custom SMARTS definitions and covalent warheads are also supported.
For medicinal and computational chemists, the pharmacophore toolbox provides a vast playground for defining the features of a series as precisely and purposefully as possible.
A Mode That Encourages Better Decisions
What makes Activity Spotter special is not just that it finds 3D patterns. It is that it does so in a way that is fast, intuitive, controllable, and immediately useful. Most importantly, it helps teams move from raw activity tables to spatially grounded hypotheses they can actually use.
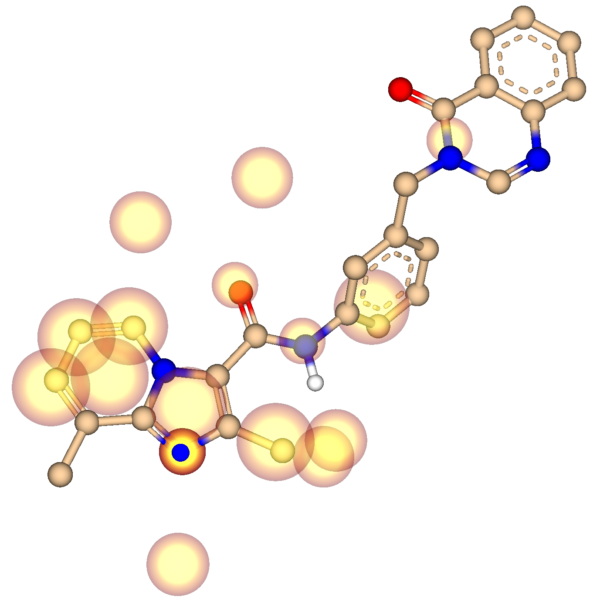
Activity spots show the user which features are particularly well differentiated among active ligands. These include hydrogen-bond partners, aromatic systems, and lipophilic groups on the ligands themselves. In addition, potential interaction spots outside the ligand cluster are displayed: these pseudo-receptor features indicate the presence of interaction partners on the target itself and can likewise be exploited through suitable pharmacophores.
- It helps medicinal chemists see why compounds succeed or fail.
- It gives computational chemists a framework they can actively shape.
- It supports mixed chemotypes, rapid iteration, and direct translation into pharmacophore-based screening.
Prioritization of the Most Informative Features
Not all spots are equally meaningful. Imagine a compound series, where each member contains a particular scaffold. Is the scaffold then really driving biological activity if it is also present in all inactive entries? In those cases, the Activity Spotter makes the distinctions visible.
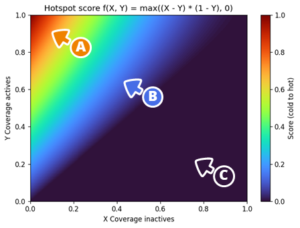
Weight principle: (A) Activity spots are those that cover many actives with few/no inactives. (B) Activity Spots featuring more inactives rank lower. (C) Inactivity Spots are regions that never contain an active. These are strong indicators of inactivity and can be treated as avoidance zones during design.
The Mode ranks spots based on how well they separate actives from inactives. Features that occur broadly among active compounds while staying absent from inactive receive a better rank. Features that appear in both inactives and actives are ranked lower. This matters because it keeps the output focused on what is truly discriminative, instead of overwhelming with undifferentiated pharmacophoric clutter. Thus, it helps crystallizing the features that best explain the observed effect pattern without boring users with redundant and trivial observations.
What Makes Activity Spotter Special?
It Work Across Chemotypes – Not Just Within a Single Series
The Mode is not restricted to a narrow congeneric series. It can use closely related compound, but can also learn from different chemotypes, as long as the molecules are brought into a meaningful common 3D frame. This is exteremely valuable for real projects: Discovery teams often do not work with a perfectly clean single-series dataset, but have to rely on mixed sources, alternative scaffolds, and contradicting hypotheses.
For medicinal and computational chemists, this means a better chance of recognizing transferabel 3D requirements beyond one single chemotype and translating this into the opportunity to test whether a shared pharmacophoric logic exists across structurally diverse matter.
A Framework for Computational Chemists They can Actively Shape
Instead of hiding the most important modeling assumption behind a black box, the mode lets computational chemists control the crucial step: how molecules are placed into a common 3D hypothesis. You can guide the alignment using your own scientific judgment, whether through template molecules, pharmacophore definitions, docking-derived poses, binding-site superposition, or multiple templates to compare alternative hypotheses.
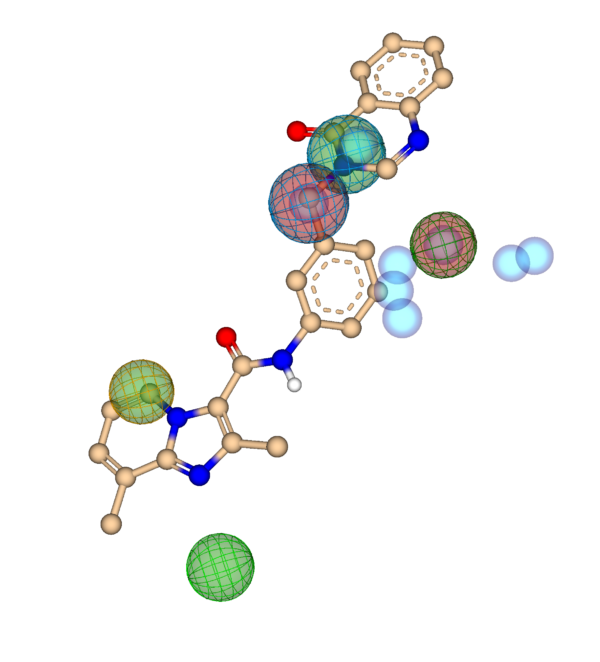
Insights from Activity and Inactivity spots help to set up meaningful pharmacophore constraints.
Extreme Speed
Activity Spotter can derive activity and inactivity features from even large sets of compounds in seconds, without forcing users through tedious preprocessing into a separate format. That speed changes how scientists can work with the method.
Instead of treating the analysis as a one-shot exercise, you can use it iteratively. You can test different activity thresholds. You can vary which compounds are considered active or inactive. You can refine a hypothesis, rerun the model, inspect the result, and immediately continue.
This is especially important because activity labels are often not as binary as they appear. In many projects, the distinction between active and inactive is a scientific choice shaped by assay context, project goals, and data confidence. Activity Spotter’s speed makes it practical to explore these alternatives rather than commit too early to a single classification.
Detecting Activity and Pseudo-Limits
Many workflows focus only on favorable features. Activity Spotter goes further by identifying both Activity Spots and Inactivity Spots. In other words, it does not just show which regions are associated with successful compounds, but also which features are consistently linked to failure.
This dual perspective is one of the most distinctive aspects of the mode. Activity Spots reveal which hydrogen-bonding features, aromatic systems, lipophilic groups, or interaction patterns are particularly well differentiated among active ligands and therefore worth preserving or strengthening. Inactivity Spots, by contrast, highlight features that repeatedly appear in inactive molecules but are absent in actives. That makes them especially valuable for understanding negative SAR.
In practical terms, these inactivity regions can often be interpreted as pseudo-limits: warning zones that may reflect steric clashes, poor fit, or electronic mismatches with the binding site. By treating them as exclusion zones during design, teams can steer optimization away from chemical dead ends and focus on modifications with a higher chance of success.
Pre-Alignment: The Basis for Meaningful Results
Activity Spotter requires an aligned set of molecules in a common 3D frame. This is essential, because the mode can only derive meaningful activity and inactivity spots when comparable features occupy comparable positions in space.
Required pre-alignment is not a drawback, but a strength: the scientist stays in control of the alignment hypothesis. Computational chemists can guide the setup using template molecules, pharmacophore definitions, docking poses, pocket alignment, or multiple templates where needed. As a best practice, it is often most effective to use a rigid compound or a known active conformation as the reference. Fewer degrees of freedom usually mean more reliable alignments and cleaner spot patterns.
The alignment itself can come from FlexS, the Similarity Scanner in SeeSAR, ligands extracted from aligned PDB structures, or other external tools. What matters is that the molecules are placed into a chemically meaningful shared 3D context.