



The latest BioSolveIT publication is now available Open Access. The paper, ‘A Benchmark Set of Bioactive Molecules for Diversity Analysis of Compound Libraries and Combinatorial Chemical Spaces,’ tackles two main themes: the creation of benchmark sets of bioactive compounds for day-to-day use in drug discovery projects, and the application of these sets to assess how well combinatorial Chemical Spaces and commercial compound libraries can deliver relevant chemistry.
Key Findings
- Chemical Spaces vs. Libraries: Chemical Spaces generally provided a greater number of compounds similar to the query molecules than the enumerated libraries and also offered unique scaffolds for each search method.
- Search Methods: All three search methods successfully identified relevant chemistry within the Chemical Spaces, with fundamental trends remaining consistent across all methods. FTrees results were the farthest from the query compound due to its pharmacophore-based approach, while SpaceLight and SpaceMACS were closer because they rely on heavy atom connectivity.
- Coverage and Blind Spots: All sources showed good coverage of classic “drug-like” structures. However, a significant blind spot was identified for more complex, hydrophilic compounds (e.g., nucleotides or those with charged groups) and natural-product-like compounds (e.g., sp3-rich carbon systems). The authors suggest this is due to the lack of available building blocks, challenging synthetic reactions, or increased reactivity of building blocks.
- Efficiency: The search algorithms performed more efficiently on the combinatorial Chemical Spaces compared to the enumerated libraries, based on the required computation time per compound.
Generation of Bioactive Compound Benchmark Sets
First, three benchmark sets were created to probe how well compound collections can return relevant compounds for hit finding and analog expansion. Starting from ~11 million ChEMBL bioactivity records, potency data was mined and a variety of filters were applied (e.g., activity < 1000 nM, MW < 800 g/mol, ≥ 10 heavy atoms; exclusions for macrocycles, off-targets, imprecise/invalid entries, duplicates, and singleton scaffolds).
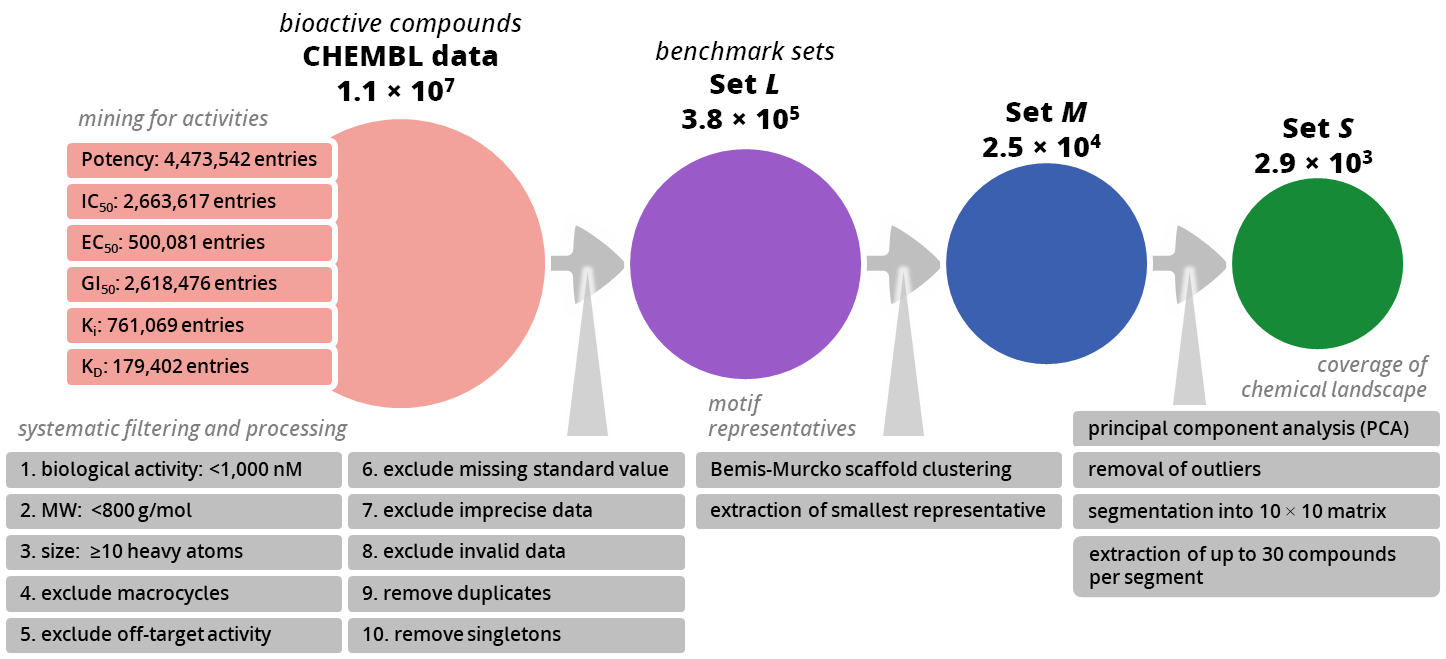
This produced:
- Set L (≈3.8 × 10⁵) — potency-filtered “motif representatives.”
- Set M (≈2.5 × 10⁴) — Bemis–Murcko scaffold clustering with the smallest member retained per scaffold.
- Set S (≈2.9 × 10³) — a PCA-balanced subset created by mapping chemical space, removing outliers, segmenting a 10×10 grid, and sampling up to 30 molecules per cell for broad, uniform coverage.
These size-tiered, ready-to-use sets formed the basis for evaluating the capacity of commercial combinatorial Chemical Spaces and enumerated libraries to supply close analogs and diverse, project-relevant chemistry.
Assessment of Commercial Chemical Spaces and Libraries
With Set S as queries, both on-demand combinatorial Chemical Spaces and enumerated were investigated to provide related compounds. Six Spaces spanning billions to trillions of make-on-demand compounds (eXplore, REAL Space, GalaXi, AMBrosia, CHEMriya, Freedom Space) and four enumerated catalogs (Mcule, Molport, Life Chemicals, ChemDiv) were screened.
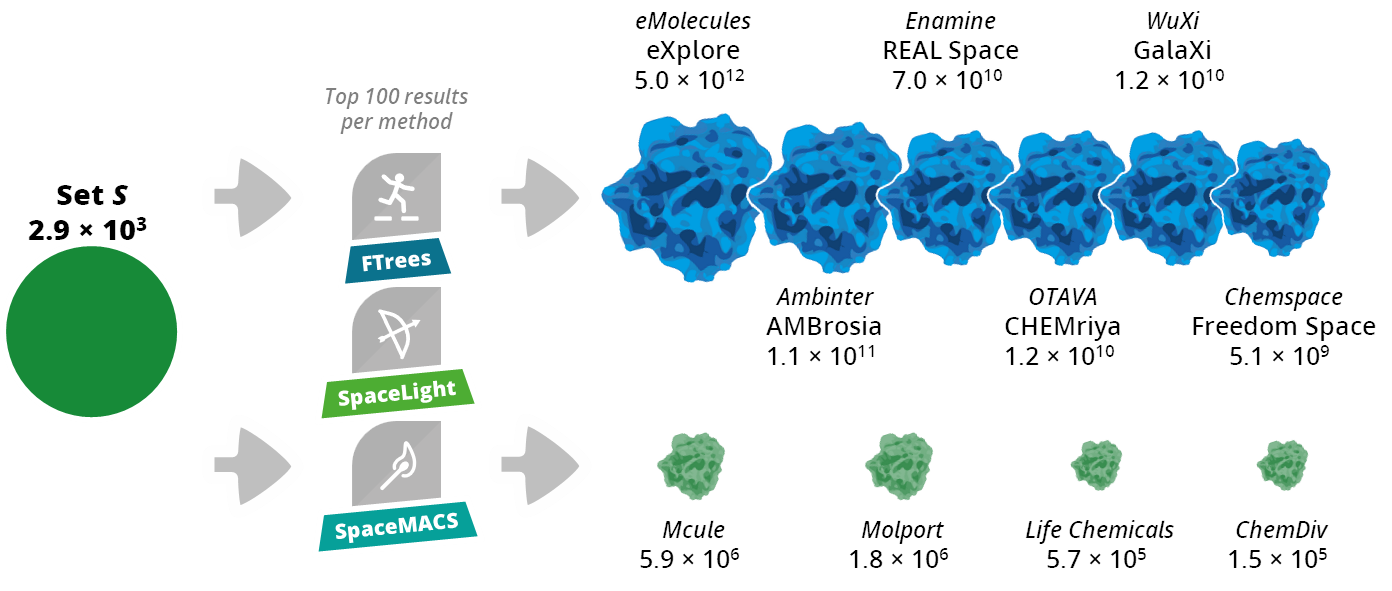
For every Set S molecule, three complementary methods (FTrees (pharmacophore features), SpaceLight (fingerprints), and SpaceMACS (maximum common substructure)) returned the top 100 hits per source. Mean similarity to the query, exact/near-exact match rates, scaffold uniqueness, and coverage across the chemical-space map (PCA quadrants) were measured.
Spaces generally yielded more and closer analogs than enumerated libraries, with eXplore and REAL Space leading among Spaces and Mcule strongest among libraries. Each method contributed distinct, often unique, scaffolds, providing flexibility for project-specific library design.
The publication then proceeds to analyze the results in detail, breaking down performance by method and by source, quantifying exact and near-exact matches, and visualizing scaffold uniqueness across suppliers. A quadrant-by-quadrant view of chemical space shows where collections excel (classic drug-like regions) and where blind spots persist (polar and bRo5-like chemistry), illustrated with concrete query examples. The study also benchmarks runtime and scale, explaining why combinatorial Spaces deliver results so quickly, and closes with practical guidance on when to favor FTrees vs. SpaceLight/SpaceMACS and how to combine sources to maximize novelty and relevance.
The full publication is freely accessible to everyone. It marks a milestone in the first holistic efforts to map chemical space and identify areas for improvement and blind spots of commercial compound sources.