


Drug Report Recap features recent scientific reports with BioSolveIT applications, platforms, and tools. We would like to highlight high-quality publications that made creative and efficient use of molecular modelling and computer-aided drug discovery.
Structure-based Virtual Screening for Inhibitors of FLT3 and MNK2
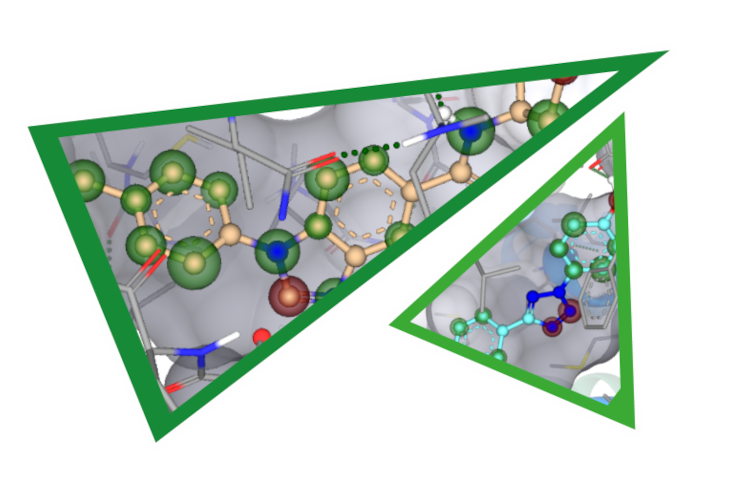
In the first drug report, Yen et al. performed structure-based virtual screening on two targets, namely Fms-like tyrosine kinase 3 (FLT3) and Mitogen-activated protein kinase (MAPK)-interacting kinase 2 (MNK2), with the aim to avoid drug resistance. Starting from two X-ray crystal structures (PDB-IDs: 4RT7 and 6CJE) they docked approximately 1.6 million compounds of the ChemDiv catalog at both targets and used interactions analysis to enhance the hit rate. After applying their workflow, they purchased 13 compounds of which three successfully inhibited FLT3 (23 % hit rate) and one that inhibited MNK2 (8% hit rate). The most promising candidate (K783-0308) displayed IC50 values of 680 and 406 nM, respectively.
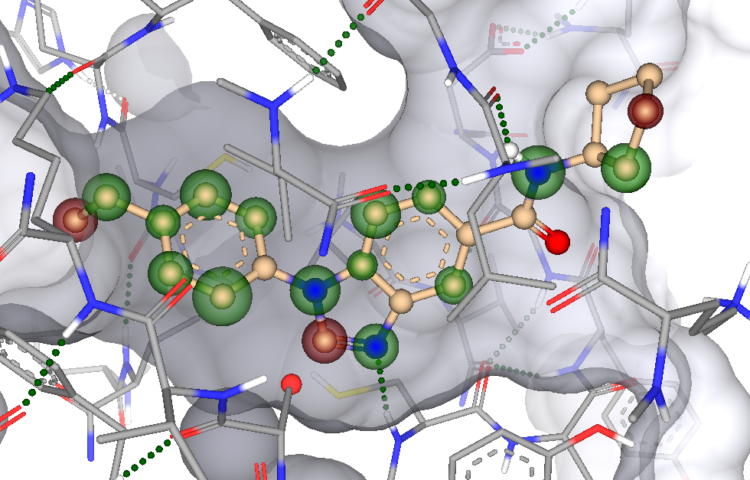
By analyzing an additional set of 13 close-analogs to compound K783-0308 they were able to elucidate important interaction features of the ligand with the receptor. Additionally, K783-0308 was screened at 82 kinases from different families and only FLT3 and MNK2 were inhibited with an inhibition percentage above 50%.
Read the full publication here.
Assessment of Tetrazole Derivatives Bearing Bisphenol Structures
Xenoestrogens are molecules that mimic endogenous estrogen and cause endocrine disruption. Gadgoli et al. started from the xenoestrogen bisphenol A (BPA) with the goal to design novel monomers for polymer chemistry to fulfil unmet market needs of safe and moldable material. This approach differs from the usual drug design process, as the group aimed to decrease the affinity towards targets (in this case estrogen-binding receptors such as androgen receptor (AR), estrogen receptor-alpha (ERα), and estrogen receptor-beta (ERβ)) compared to the common approach of binding affinity improvement.
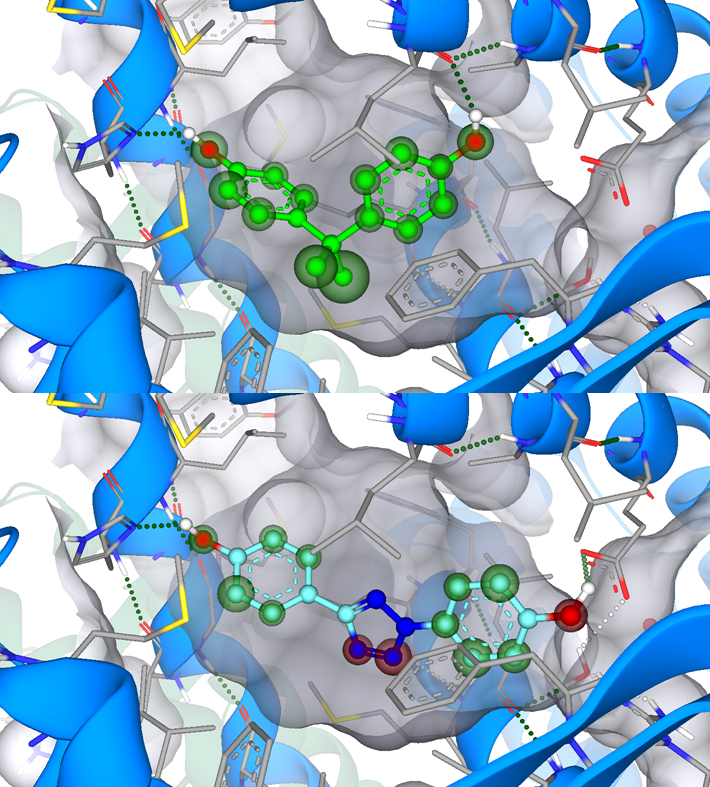
Using ReCore in SeeSAR within their workflow, they screened for novel scaffolds as replacement for the isopropylidene group of BPA. Subsequently, the designed compounds were docked, scored with Hyde, and assessed for their binding affinity at the targets. Promising candidates were synthesized and pharmacologically assessed for their receptor activity. The tetrazole scaffold displayed decreased binding affinity. Furthermore, introduction of methyl and methoxy groups at the phenyl rings further decreased the inhibitory potency of the compounds almost abolishing antagonism.
Read the full publication here.
Did you get interested in drug discovery with BioSolveIT applications?
Download SeeSAR and infiniSee and enhance your drug discovery process.