


In modern drug discovery, speed is trumps because time is money. Virtual screening methods bring those benefits to the table that ring a bell for any project leader: They are cheap and fast.
In the past, several FDA-approved drugs emerged from computer-aided structure-based drug discovery. Among the used methods, molecular docking has become one of the most popular and helpful tools for lead identification, SAR analysis, and more. Yet, it is not really handy anymore with the advent and successes of purchasable Chemical Spaces that exceed 1010 molecular members.
But there is remedy now.
Virtual screening and its Limits
Roger Sayle of NextMove once said “Docking is like a discotheque, it’s all about posing and scoring.” Indeed! Docking methods are used to predict the “best”, aka the most likely, binding mode of a ligand in a target; then they score, for example, using sums of physically motivated energy descriptions.
With static targets, the pose generation process is usually limited by ligand torsional sampling. This dictates the required calculation time. However, even leaving the target static, a computation of thousands of compounds is limited for most labs in the region of 108 molecules. Simulations that involve motions of the target require even much larger computational resources, often resulting in day-long computations for a single ligand.
With the expansion of accessible and purchasable molecules well beyond the count of 109 conventional docking methods fail to meet the demands of fast and easy-to-perform virtual screening campaigns. Here, the user gets confronted with a novel “luxury problem” – the unlimited accessibility of potential hit molecules overstrains the most powerful docking tool. Virtual screening campaigns involving only a few millions of molecules that take days-to-weeks would take ten to hundreds of years to be performed on a normal working station.
FTrees – Fuzzy Similarities at the Speed of Light
Our FTrees application addresses this problem. It kicks the door open to a highly efficient scan of the purchasable space the output of which is there in minutes and can then be used for 3D methods.
FTrees (short for Feature Trees) analyzes the overall topology and fuzzy pharmacophore properties of the molecule and translates the data into so-called molecular descriptors. With these descriptors, FTrees swiftly navigates through compound libraries and Chemical Spaces looking for molecules with similar features. Several drug discovery campaigns have demonstrated the capabilities of FTrees to screen ultra-large Chemical Spaces where conventional molecular docking would certainly not cope any longer.
The main reason for that is that FTrees works at unprecedented speed. But how fast is FTrees actually?
We took a look at the performance of virtual high-throughput screening (vHTS) first. In an exemplary standard virtual screening campaign in which 3,000,000 molecules were docked within five days (432,000 seconds), the effective docking time came out at 0.144 seconds per compound.
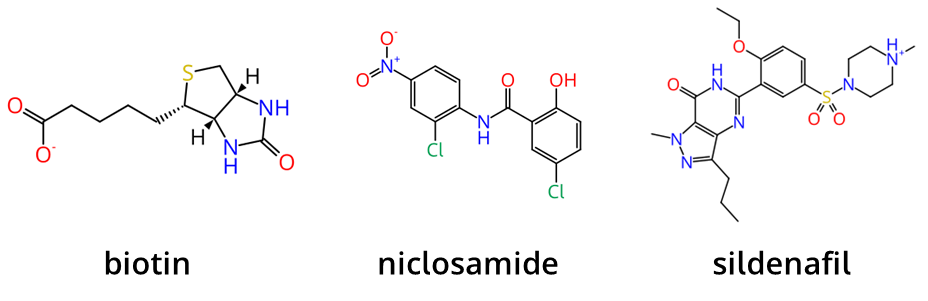
To capture the speed of FTrees we performed a screening on enumerated and combinatorial data sets. Our evaluation set consisted of three well-known molecules with varying complexity: biotin (243.3 g/mol), niclosamide (327.1 g/mol), sildenafil (475.6 g/mol). Used parameters and hardware are below.
For the enumerated data set, FTrees performed 100-1.000 faster than conventional docking. The difference in performance becomes even more evident when FTrees is applied on combinatorial Chemical Spaces: the exploration of 2.1 x 109 and 1.5 x 1010 compounds was 10 to 100 million-times faster than standard docking, respectively. Pros and cons of combinatorial Chemical Spaces vs. enumerated libraries has been previously discussed.
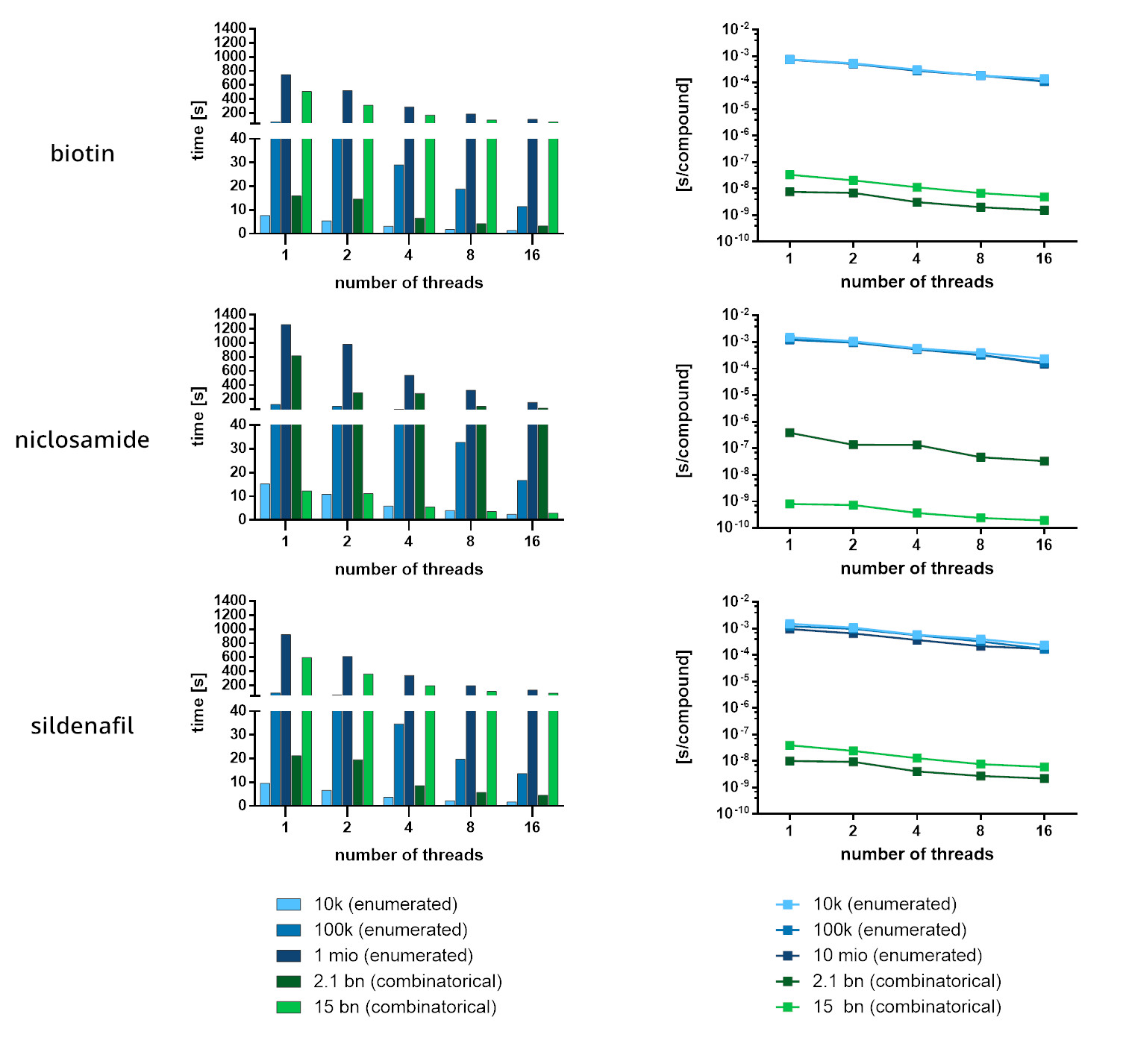
Furthermore, more query compounds accelerate the navigation through combinatorical spaces. Once the space is build-up with the first search, FTrees can take advantage of that for subsequent queries. It is worthwhile to mention that FTrees, by design of the search algorithm, does not discard molecules in a manner of filtering during the process but explores the entire set of compounds in the process, thus nothing will be missed that shouldn’t be missed.
While docking has not yet reached the point of handling ultra-large Chemical Spaces, FTrees elegantly complements the drug discovery process as a leading hand. It opens the door to a vast ocean of purchasable compounds or can be used pre-screen as a filter. So next time before you dock, expand your possibilies and do not miss potential hits.
FTrees is a key part of our software platform infiniSee.
Methods
- FTrees settings
- -i x.sdf -s y.sdf/.space –o z.sdf –maxNofResults 100 –minSimilarityThreshold 0.8 –targetSimilarity 1 –totalDiversity 1 -–thread-count 1/2/4/8/16
- Hardware
- Intel(R) Xeon(R) W-3223 CPU @ 3.50GHz, 8 cores (16 threads), openSUSE 15.1
- enumerated input data set
- eMolecules 2,344,946 molecules as supplied with FragXplorer workflow (10k, 100k, and 1 mio molecules as sd-file)
- combinatorial input data set
- REALSpace_2020-11.space
- 2.1bn-GalaXi_2020-11.space