With infiniSee 7.0 and infiniSee xREAL 7.0, a new database (DB) format for Chemical Spaces is being introduced.
Going forward, all Space updates will be released in this new format. Therefore, we recommend updating to the latest versions of the platforms and the associated command-line tools.
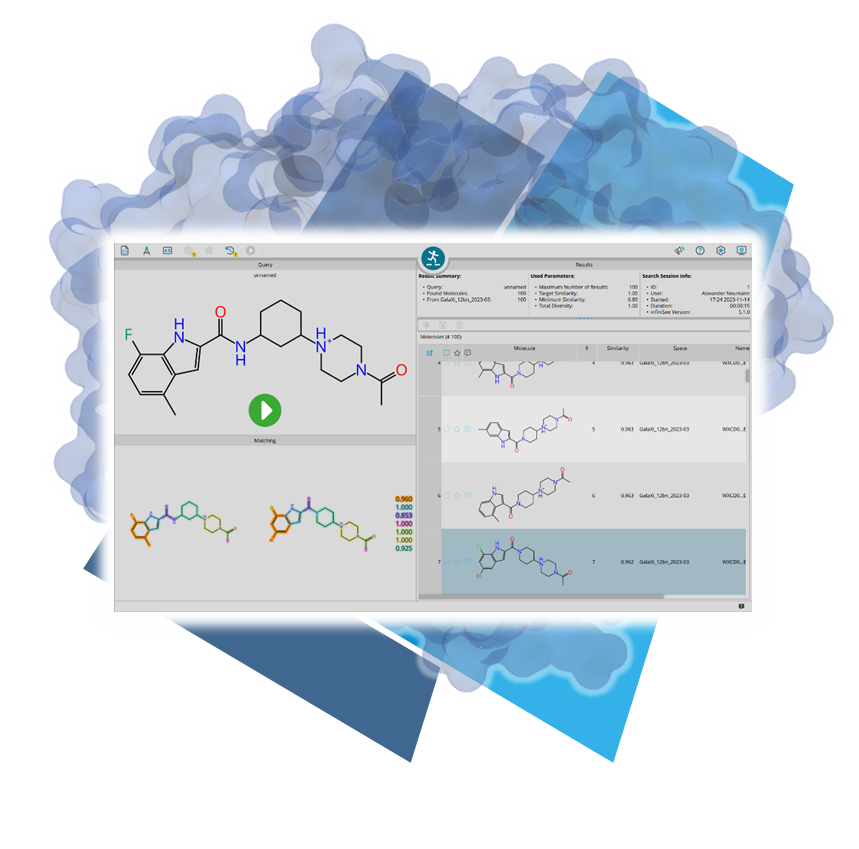
A major advantage of the new format is that it enables significantly faster screenings and reduces RAM usage.
Please note: The new format is not backward-compatible.
If you require older versions of the Chemical Spaces, feel free to contact our
support team.
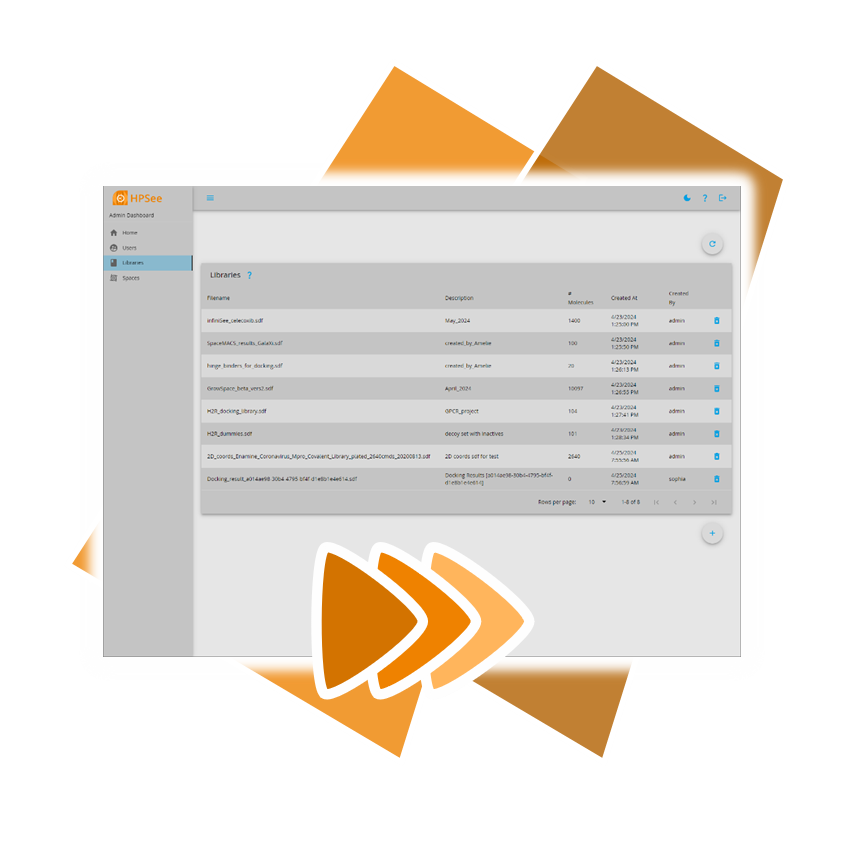
You can download the new versions of the Chemical Spaces
following this link.