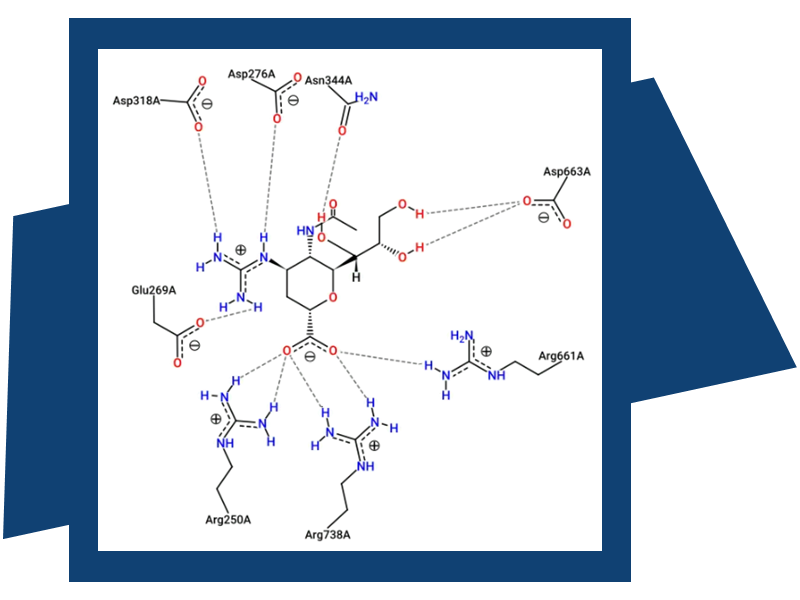
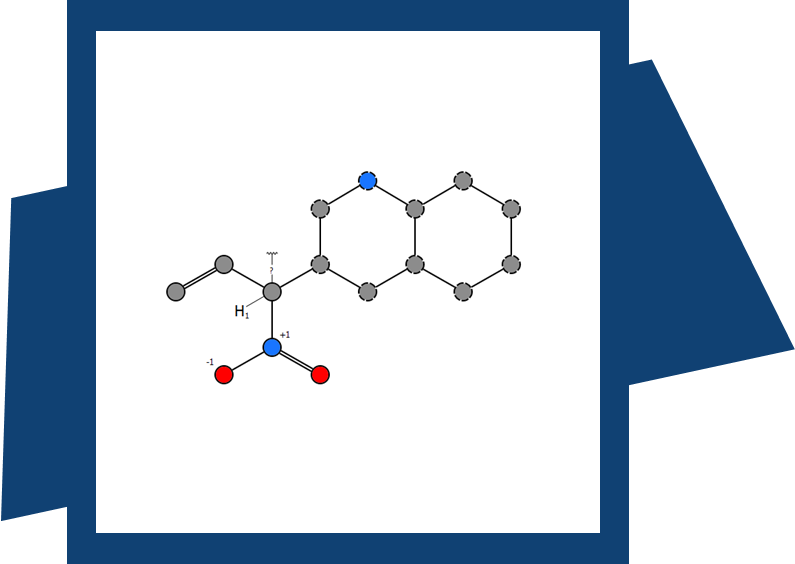
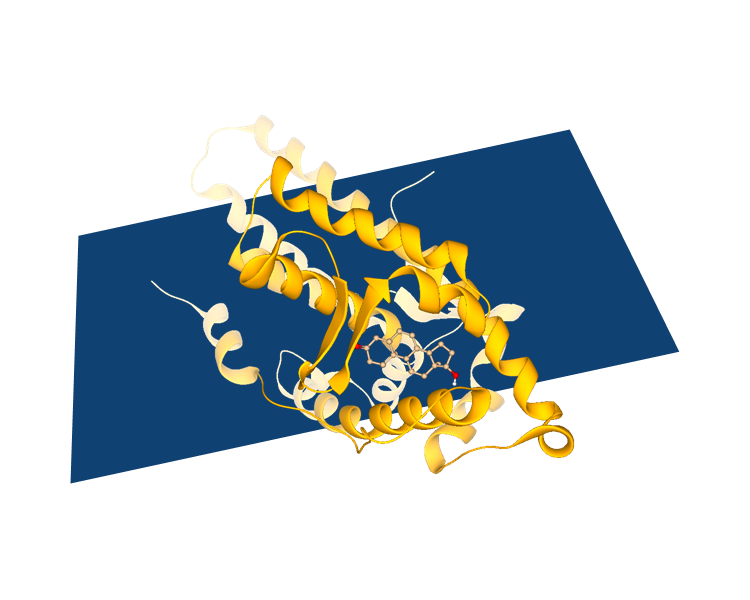
SeeSAR
Drug Design Dashboard








* Important notice
No support is granted and any warranty or liability is hereby expressly excluded. Proceed at your own risk!
Please acknowledge scientific ownership. Any publication in reference to these downloads must cite the respective original publication.
Please provide feedback.